Supplementary information: Test methods used during development or manufacture: Recommendations for quality requirements when plant-derived artemisinin is used as a starting material in the production of antimalarial active pharmaceutical ingredients 1
1 WHO Expert Committee on Specifications for Pharmaceutical Preparations, Forty-sixth report. Geneva, World Health Organization, 2012, Annex 6 (WHO Technical Report Series, No. 970).
1. Introduction
The harmonized good manufacturing practices (GMP) (1,2) describe requirements for the production of active pharmaceutical ingredients (APIs). The applicability of these requirements begins with a defined starting material as follows:
“An API starting material is a raw material, intermediate or an API that is used in the production and that is incorporated as a significant structural fragment into the structure of the API. An API starting material can be an article of commerce, a material purchased from one or more suppliers under contract or commercial agreement or produced in-house. API starting materials normally have defined chemical properties and structure.”
The focus of GMP for APIs is for field inspector use rather than in applications for marketing authorization. It defines what may be considered as a starting material and provides guidance on where GMP is applied. The GMP guidelines do not apply to steps taken prior to the first introduction of the defined starting material. The manufacturer should designate and document the rationale for the point at which production of the API begins. For a synthesis process this is known as the point at which the starting materials are entered into processes.
From a regulatory standpoint the use of API starting materials marks the beginning of the detailed description of the process. The applicant for marketing authorization should propose and justify which substance should be considered as the API starting material, e.g. incorporated as a significant structural fragment into the structure of the active substance.
In practice the designation of a starting material may be difficult. The number of steps separating the starting material from the final API is an issue to be decided on a case-by-case basis, subject to the manufacturer’s proposal and assessors’ evaluation. Since a designated starting material may be obtained from multiple sources, it is necessary to have well-defined quality requirements to ensure that the APIs produced meet specifications. Establishing these requirements may involve a compromise between the desire for a pure starting material and the impact of this on cost of API production. Impurities can be tolerated in the starting material if the API manufacturing process has been shown to efficiently remove them. Redundant purification steps may reduce the yield of the final API and thus further increase its cost.
Artemisinin derivatives used in artemisinin-based combination therapy (ACT) are synthesized from artemisinin in one or two synthetic steps. Artemisinin is typically produced as an isolate from Artemisia annua L. Artemisinin complies with the definition of a “starting material”, as defined above and described in certain national, regional and international guidelines. It is:
- a material used in the production of the API that is incorporated into the API as a significant structural element;
- commercially available;
- a compound whose name, chemical structure, chemical and physical characteristics, properties and impurity profile are well defined;
- obtained by commonly known procedures.
As artemisinin is extracted from plant material and prior intermediates are thus not available it is logical to designate this compound as the starting material for its derivatives.
A monograph appears in The International Pharmacopoeia for artemisinin used as an API. However, at present, artemisinin is mainly used as a starting material for artemisinin-derived APIs and not as an API.
The level of quality of the artemisinin should be acceptable for its intended use as the starting material for the production of artemisinin derivatives. The specifications presented below take into account an acceptable balance of benefit versus risk between the quality of artemisinin used as a starting material and the quality required for artemisinin derivatives for use as APIs.
However, competent authorities may accept other impurity profile levels depending on the capability of the manufacturing process to lead to artemisinin-derived APIs at least compliant with the relevant monographs of The International Pharmacopoeia.
The purpose of this document is to offer a global approach to defining the level of quality requirements of artemisinin when used as a starting material for the production of its API derivatives used in ACT formulations. It does not apply to cases where artemisinin is used as an API. It is intended that the recommendations for requirements outlined in this document will apply to artemisinin extracted from Artemisia annua L. regardless of variations in agricultural environment or variations in extraction and purification steps. In addition, in order to ensure appropriate quality of the derived APIs, the manufacturer may add additional tests, such as tests for residual solvents and heavy metals, among others, and/or require tighter specifications. For artemisinin produced using synthetic chemical processes or by fermentation other requirements may be applicable.
2. Characterization of artemisinin
Provided that artemisinin intended for use as a starting material has been correctly identified the major quality concern is the presence and level of impurities with the potential to affect the purity of subsequent API derivatives. Impurities may originate from the plant extracts or arise from the purification process or from degradation. Different biosynthetic routes may be used at different stages in the plant’s development and there are claims of variability between growing regions and environments. Despite a lack of consensus on a single biosynthetic route several potential impurities are common to different routes. These include artemisinic acid, dihydroartemisinic acid, arteannuin B and artemisitene. Of these only artemisitene has been reported in isolated artemisinin. Recent work (3, 4) has contributed towards a clearer understanding of existing impurities and their analysis.
Examination of a wide variety of artemisinin samples produced in various regions indicated the consistent presence of two impurities: artemisitene and an artemisinin diastereomer with the stereochemistry inverted at C-9 (9-epi-artemisinin). A possible concern is that artemisinin impurities may not be detected with high-performance liquid chromatography analysis using ultraviolet detection, as used in the majority of testing laboratories. Recent work ( 5) using more sensitive general detection by mass spectrometry, however, demonstrated that additional impurities occur only in trace amounts. Isolated artemisinin is very stable. The potential degradants proposed on the basis of mechanistic studies do not occur at temperatures below 100 °C. These degradants are not observed in isolated artemisinin.
In the chemical conversion of the artemisinin starting material to its API derivatives (e.g. artesunate), the artemisinin diastereomeric impurity may be converted to a corresponding diastereomer at the C-9 position in the API derivative. However, these resulting diastereomers have not been observed in isolated APIs. The fate of artemisitene is less clear as it may be converted to the same intermediate as artemisinin.
Artemisitene-derived impurities have not been observed in artemisinin derivative APIs. Proposed limits for these impurities are based on historical results. The specifications for artemisinin starting material are based on experience with artemether and artesunate. For a new artemisinin-derived API the suitability of the specifications to control potential impurities arising during its synthesis should be demonstrated.
As the artemisinin extraction processes use solvents like dichloromethane, chloroform, ether and others, residual solvents should be indicated on the certificate of analysis issued by the supplier.
3. Tests and specifications for artemisinin starting material
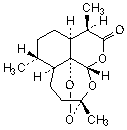
C15H22O5
Relative molecular mass: 282.3
Chemical name: (3R,5aS,6R,8aS,9R,12S,12aR)-3,6,9-trimethyloctahydro-3,12-epoxypyrano[4,3-j]-1,2-benzodioxepin-10(3H)-one; CAS Reg. No. 63968-64-9.
Description: Colourless needles or a white to almost white to slightly yellow, crystalline powder.
Category: Starting material for the synthesis of artemisinin derivative APIs.
Storage : Artemisinin should be kept in a well-closed container, protected from light.
Requirements
Artemisinin contains not less than 95.0% and not more than the equivalent of 102.0% of C15H22O5 calculated with reference to the dried substance.
Identity tests
Carry out the examination as described under 1.7 Spectrophotometry in the infrared region of The International Pharmacopoeia (6 ). The infrared absorption spectrum is concordant with the spectrum obtained from artemisinin RS or with the reference spectrum of artemisinin in The International Pharmacopoeia.
Specific optical rotation. Use a 10 mg/mL solution in dehydrated ethanol R;
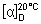 = + 75° to +78°
= + 75° to +78°
Loss on drying. Dry to constant mass at 80 °C; it loses not more than 10.0 mg/g.
Related substances
Note : It may be possible to justify other limits when artemisinin as a starting material is used in a particular synthesis and manufacturing process, by validation of the levels and limits of the impurities in the final API.
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography of The International Pharmacopoeia (6). Use the chromatographic conditions and prepare solutions (1) and (2) as described below under Assay. For solution (3) dilute 1 mL of solution (1) to 100 mL with the mobile phase.
Inject separately 20 μl of solutions (1), (2) and (3). Record the chromatograms for about 1.5 times the retention time of artemisinin. In the chromatogram obtained with solution (2) artemisitene (impurity A) is eluted at the relative retention of about 0.79 with reference to artemisinin (retention time about 10 minutes). The test is not valid unless the resolution between the peak of artemisitene and the peak of artemisinin is at least 4. The chromatogram obtained with solution (1) may show a peak due to impurity B eluting at a retention of about 0.85 with reference to artemisinin.
In the chromatogram obtained with solution (1):
- the area of any peak corresponding to impurity A, when multiplied by a correction factor of 0.027, is not greater than 0.2 times the area of the peak in the chromatogram obtained with solution (3) (0.2%);
- the area of any peak corresponding to impurity B is not greater than the area of the peak in the chromatogram obtained with solution (3) (1.0%);
- the area of any peak other than the principal peak is not greater than 0.5 times the area of the peak in the chromatogram obtained with solution (3) (0.5%);
- the sum of the corrected area of any peak corresponding to impurity A and the areas of all the peaks, apart from the principal peak, is not greater than 3 times the area of the peak obtained with solution (3) (3.0%). Disregard any peak with an area less than 0.1 times the area of the principal peak obtained with solution (3) (0.1%).
Assay
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography of The International Pharmacopoeia (6) using a stainless steel column (15 cm × 4.6 mm) packed with 5 μm particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups. The mobile phase consists of a 50:50 mixture of acetonitrile and water, pumped at a flow rate of 1.0 mL/minute. As a detector use an ultraviolet spectrophotometer set at a wavelength of 210 nm.
Prepare the following solutions. For solution (1) prepare a 5.0 mg/mL solution of the test substance in the mobile phase. For solution (2) prepare a 5.0 mg/mL solution of artemisinin RS (containing artemisinin and impurity A) in the mobile phase.
Inject separately 20 μl of solutions (1) and (2). Record the chromatograms for about 1.5 times the retention time of artemisinin. In the chromatogram obtained with solution (2) artemisitene (impurity A) is eluted at the relative retention of 0.79 with reference to artemisinin (retention time about 10 minutes). The test is not valid unless the resolution between the peak of artemisitene and the peak of artemisinin is at least 4. The chromatogram obtained with solution (1) may show a peak due to impurity B eluting at a retention of about 0.85 with reference to artemisinin.
Measure the areas of the peak responses obtained in the chromatograms from solutions (1) and (2), and calculate the content of C15H22O5 with reference to the dried substance.
Impurities
A.
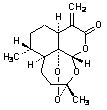
(3R,5aS,6R,8aS,12S,12aR)-3,6-dimethyl-9-methylideneoctahydro-3,12-epoxypyrano[4,3-j ]-1,2-benzodioxepin-10(3H)-one (artemisitene)
B.
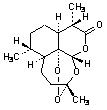
(3R,5aS,6R,8aS,9S,12S,12aR)-3,6,9-trimethyloctahydro-3,12-epoxypyrano[4,3-j ]-1,2-benzodioxepin-10(3H)-one (9-epi-artemisinin)
References
- WHO good manufacturing practices for active pharmaceutical ingredients. In: WHO Expert Committee on Specifications for Pharmaceutical Preparations. Forty-fourth report. Geneva, World Health Organization (WHO Technical Report Series, No. 957), Annex 2, 2010.
- International Conference on Harmonisation (ICH) Topic Q7: Note for guidance on good manufacturing practice for active pharmaceutical ingredients. London, EMEA, 2000 (CPMP/ICH/4106/00); http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002825.pdf..
- Lapkin AA et al. Development of HPLC analytical protocols for quantification of artemisinin in biomass and extracts. Journal of Pharmaceutical and Biomedical Analysis, 2009, 49: 908–915.
- Stringham RW et al. High performance liquid chromatographic evaluation of artemisinin, raw material in the synthesis of artesunate and artemether. Journal of Chromatography A, 2009, 1216: 8918–8925.
- Stringham RW et al. Verification of the identities of impurities in artemisinin and correction of their elution order in high performance liquid chromatography. Journal of Chromatography A, 2011, 1218: 6838–6842.
- The International Pharmacopoeia, 4th ed., Vol. 1: General notices; monographs for pharmaceutical substances (A–O) and Vol. 2: Monographs for pharmaceutical substances (P–Z); monographs for dosage forms and radiopharmaceutical preparations; methods of analysis; reagents. Geneva, World Health Organization, 2006 online; The International Pharmacopoeia, 4th ed., First, Second and Third Supplements, 2013, available on CD-ROM.