Monographs: Pharmaceutical substances: Artesunate (Artesunatum)

C19H28O8
Relative molecular mass. 384.4.
Chemical names. (3R,5aS,6R,8aS,9R,10S,12R,12aR)-3,6,9-trimethyldecahydro-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10-yl hydrogen butanedioate; (3R,5aS,6R,8aS,9R,10S,12R,12aR)-decahydro-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10-ol, hydrogen succinate; CAS Reg. No. 88495-63-0.
Description. A fine, white crystalline powder.
Solubility. Very slightly soluble in water; very soluble in dichloromethane R; freely soluble in ethanol (~750 g/l) TS and acetone R.
Category. Antimalarial.
Storage. Artesunate should be kept in a well-closed container or, if sterile, in a hermetically closed container. It should be protected from light.
Labelling. The label states where applicable:
– that the substance is free from bacterial endotoxins;
– that the substance is sterile.
Requirements
Definition. Artesunate contains not less than 96.0% and not more than 102.0% of artesunate (C19H28O8) using Assay method A, and not less than 98.0% and not more than 102.0% of artesunate (C19H28O8) using Assay method B, both calculated with reference to the anhydrous substance.
Identity tests
•Either test A alone or tests B, C and D may be applied.
A. Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from artesunate RS or with the reference spectrum of artesunate.
B. Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica gel R5 as the coating substance and a mixture of 70 volumes of ethanol R, 30 volumes of toluene R and 1.5 volumes of ammonia (~260 g/l) TS as the mobile phase. Apply separately to the plate 1 μl of the following two solutions in methanol R. For solution (A) use 1.0 mg of the test substance per mL. For solution (B) use 1.0 mg of artesunate RS per mL. After removing the plate from the chromatographic chamber allow it to dry in air or in a current of cool air. Spray with anisaldehyde/methanol TS and heat the plate to 120 °C for 5 minutes. Examine the chromatogram in daylight.
The principal spot obtained with solution (A) corresponds in position, appearance and intensity with that obtained with solution (B).
C. Dissolve 0.1 g in 40 mL of dehydrated ethanol R, shake and filter. To half of the filtrate (keep the remaining filtrate for test D) add about 0.5 mL of hydroxylamine hydrochloride TS2 and 0.25 mL of sodium hydroxide (~80 g/l) TS. Heat the mixture in a water-bath to boiling, cool, add 5 drops of hydrochloric acid (~70 g/l) TS and 2 drops of ferric chloride (50 g/l) TS; a reddish-brown colour is produced.
D. Evaporate the remaining filtrate from test C on a water-bath to a volume of about 5 mL. Place a few drops of the mixture on a white porcelain dish, add one drop of vanillin/sulfuric acid TS1; a red colour is produced.
Specific optical rotation. Use a 10 mg/mL solution in dichloromethane R and calculate with reference to the anhydrous substance;
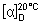 = +4.5° to + 6.5°.
= +4.5° to + 6.5°.
Heavy metals. Use 1.0 g for the preparation of the test solution as described under 2.2.3 Limit test for heavy metals, Procedure 3; determine the heavy metals content according to Method A; not more than 20 μg/g.
Sulfated ash. (2.3) Not more than 1.0 mg/g.
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A, using 2 g of the test substance; the water content is not more than 5 mg/g.
pH value. (1.13) pH of a 10 mg/g aqueous suspension, 3.5–4.5.
Bacterial endotoxins. If intended for use in the manufacture of a parenteral dosage form without a further appropriate procedure for the removal of bacterial endotoxins, carry out the test as described under 3.4 Test for bacterial endotoxins; contains not more than 2.5 IU of endotoxin per mg.
Sterility. If intended for use in the manufacture of a parenteral dosage form without a further appropriate sterilization procedure, complies with 3.2 Test for sterility.
Related substances
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using the conditions given below under Assay method A.
Use solutions (1) and (3) as described under Assay method A. For solution (4) dilute 1 mL of solution (1) to 100 mL with acetonitrile R.
Inject separately 20 μl each of solutions (1), (3) and (4). Record the chromatograms for about 4 times the retention time of artesunate.
In the chromatogram obtained with solution (3) the following peaks are eluted at the following relative retention with reference to artesunate (retention time about 9 minutes): 10-epi-artenimol about 0.58; artenimol about 0.91; and impurity B (artemisinin) about 1.30. The test is not valid unless the peak-to-valley ratio (Hp/Hv) is at least 5.0, where Hp is the height above the extrapolated baseline of the peak due to artenimol and Hv is the height above the extrapolated baseline at the lowest point of the curve separating the peak due to artenimol from the peak due to artesunate. The chromatogram obtained with solution (1) may show a peak due to impurity C eluting at a relative retention of about 2.7 with reference to artesunate.
In the chromatogram obtained with solution (1):
– the combined areas of any peaks corresponding to 10-epi-artenimol and artenimol (impurity A) are not greater than the area of the principal peak obtained with solution (4) (1.0%);
– the area of any peak corresponding to impurity B (artemisinin) is not greater than 0.5 times the area of the principal peak obtained with solution (4) (0.5%);
– the area of any peak corresponding to impurity C, when multiplied by a correction factor of 0.07, is not greater than 0.2 times the area of the principal peak obtained with solution (4) (0.2%);
– the area of any other peak, other than the principal peak, is not greater than 0.2 times the area of the principal peak in the chromatogram obtained with solution (4) (0.2%);
– the sum of the corrected area of any peak corresponding to impurity C and the areas of all other peaks, other than the principal peak, is not greater than twice the area of the principal peak obtained with solution (4) (2.0%). Disregard any peak with an area less than 0.05 times the area of the principal peak in the chromatogram obtained with solution (4) (0.05%).
Assay
• Either method A or method B may be applied.
A. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography , using a stainless steel column (10 cm × 4.6 mm) packed with particles of silica gel, the surface of which has been modified with chemically bonded octadecylsilyl groups (3 μm). As the mobile phase use a mixture of 44 volumes of acetonitrile R and 56 volumes of buffer pH 3.0
Prepare the buffer pH 3.0 by dissolving 1.36 g of potassium dihydrogen phosphate R in 900 mL of water R, adjust the pH to 3.0 with phosphoric acid (~1440 g/l) TS and dilute to 1000 mL with water R.
Prepare the following solutions in acetonitrile R. For solution (1) dissolve 40 mg of the test substance, accurately weighed and dilute to 10 mL. For solution (2) dissolve 40 mg of artesunate RS, accurately weighed, and dilute to 10 mL. For solution (3) dissolve about 1 mg of artenimol RS, about 1 mg of artemisinin RS and about 10 mg of artesunate RS in 10 mL.
Operate with a flow rate of 1.0 mL per minute. Maintain the column temperature at 30 °C and use as detector an ultraviolet spectrophotometer set at a wavelength of about 216 nm.
Inject separately 20 μl each of solutions (1), (2) and (3). Record the chromatograms for about 4 times the retention time of artesunate. In the chromatogram obtained with solution (3) the following peaks are eluted at the following relative retention with reference to artesunate (retention time about 9 minutes): 10-epi-artenimol about 0.58, artenimol about 0.91; and impurity B (artemisinin) about 1.30. The test is not valid unless the peak-to-valley ratio (Hp/Hv) is at least 5.0, where Hp is the height above the extrapolated baseline of the peak due to artenimol and Hv is the height above the extrapolated baseline at the lowest point of the curve separating the peak due to artenimol from the peak due to artesunate. The chromatogram obtained with solution (1) may show a peak due to impurity C eluting at a relative retention of about 2.7 with reference to artesunate.
Measure the areas of the peak responses obtained in the chromatograms from solutions (1) and (2) and calculate the percentage content of artesunate (C19H28O8) with reference to the anhydrous substance, using the declared content of C19H28O8 in artesunate RS.
B. Dissolve about 0.25 g of Artesunate, accurately weighed, in 25 mL of neutralized ethanol TS and titrate with sodium hydroxide (0.05 mol/l) VS, using 2 drops of phenolphthalein/ethanol TS as indicator.
Each mL of sodium hydroxide (0.05 mol/l) VS is equivalent to 19.22 mg of C19H28O8.
Impurities
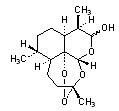
A. (3R,5aS,6R,8aS,9R,12R,12aR)-3,6,9-trimethyldecahydro-12H-3,12-epoxypyrano[4,3-j]-1,2-benzodioxepin-10-ol (dihydroartemisinins: artenimol and (10R)-artenimol),
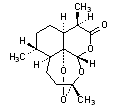
B. (3R,5aS,6R,8aS,9R,12S,12aR)-3,6,9-trimethyloctahydro-3,12-epoxypyrano[4,3-j]-1,2-benzodioxepin-10(3H)-one (artemisinin),
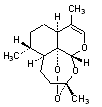
C. (3R,5aS,6R,8aS,12R,12aR)-3,6,9-trimethyl-3,4,5,5a,6,7,8,8a-octahydro-12H-3,12-epoxypyrano[4,3-j]-1,2-benzodioxepine (anhydrodihydroartemisinin).