Monographs: Pharmaceutical substances: Tenofovir disoproxil fumarate (Tenofoviri disoproxili fumaras)
Graphic formula
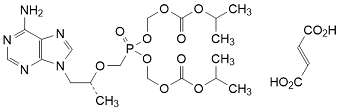
Molecular formula. C19H30N5O10P . C4H4O4
Relative molecular mass. 635.5.
Chemical names. 1,1'-bis(propan-2-yl) 1,1'-[({[(2R)-1-(6-amino-9H-purin-9-yl)-1-propan-2-yloxy]methyl}phosphonoyldioxy)dimethyl] dicarbonate (ester) hydrogen (2E)-but-2-enedioate (salt); bis(1-methylethyl) 5-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate (ester) hydrogen (2E)-but-2-enedioate (salt); bis({[(1-methylethoxy)carbonyl]oxy}methyl) {[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}phosphonate (ester) hydrogen (2E)-but-2-enedioate (salt); CAS Reg. No. 202138 50 9
Description. White to almost-white, crystalline powder.
Solubility. Slightly soluble in water R, soluble in methanol R, very slightly soluble in dichloromethane R.
Category. Antiretroviral (Nucleotide Reverse Transcriptase Inhibitor).
Storage. Tenofovir disoproxil fumarate should be kept in a tightly closed container, protected from light and stored at a temperature between 2–8 °C.
Additional information. Tenofovir disoproxil fumarate may exhibit polymorphism.
Requirements
Definition. Tenofovir disoproxil fumarate contains not less than 98.5% and not more than 101.0% of tenofovir disoproxil fumarate (C19H30N5O10P.C4H4O4), calculated with reference to the anhydrous substance.
Manufacture. The production method is validated to ensure that the substance, if tested, would comply with the following limits for the following mutagenic impurities using suitable methods:
-
not more than 5 ppm for 9-propenyladenine; and
-
not more than 50 ppm for chloromethyl isopropyl carbonate.
Identity tests
-
Either tests A and C, or tests B, E and C, or tests D, E and C may be applied.
-
Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from tenofovir disoproxil fumarate RS or with the reference spectrum of tenofovir disoproxil fumarate.
If the spectra thus obtained are not concordant, repeat the test using the residues obtained by separately dissolving the test substance and tenofovir disoproxil fumarate RS in a small amount of methanol R and evaporating to dryness. The infrared absorption spectrum is concordant with the spectrum obtained from tenofovir disoproxil fumarate RS.
-
Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica gel R6 as the coating substance and a mixture of 67 volumes of dichloromethane R, 20 volumes of acetonitrile R, 10 volumes of methanol R and 3 volumes of ammonia (~260 g/L) TS as the mobile phase. Apply separately to the plate, 5 μL of each of two solutions in methanol containing (A) 10 mg of the test substance per mL and (B) 10 mg of tenofovir disoproxil fumarate RS per mL. After removing the plate from the chromatographic chamber, allow it to dry exhaustively in air or in a current of air. Examine the plate in ultraviolet light (254 nm).
The principal spot obtained with solution (A) corresponds in position, appearance and intensity with that obtained with solution (B).
-
Carry out test C.1 or test C.2.
C.1 Determine the specific optical rotation (1.4) using a freshly prepared 10.0 mg/mL solution of the test substance in hydrochloric acid (~3.65 g/L) TS. Perform the test without delay and calculate with reference to the anhydrous substance;
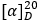 = -15 to -20.
= -15 to -20.C.2 Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using the conditions given under "Impurity G". Prepare the following solutions using a mixture of 9 volumes of dehydrated ethanol R and 1 volume of acetonitrile (v/v) as the diluent. For solution (1), dissolve 2 mg of the test substance in 20 mL. For solution (2), dissolve the content of a vial of tenofovir disoproxil fumarate for enantiomer identification RS in 0.5 mL. The retention time of the principal peak in the chromatogram obtained with solution (1) corresponds to the retention time of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (2).
-
The absorption spectrum (1.6) of a 25 µg/mL solution of the test substance in water R, when observed between 220 nm and 320 nm, exhibits a maximum at about 261 nm.
Alternatively, in combination with identity test C.2, where a diode array detector is available, record the UV spectra of the principal peaks in the chromatograms with a diode array detector in the range of 220 nm to 320 nm. The UV spectrum of the principal peak in the chromatogram obtained with solution (1) corresponds to the UV spectrum of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (2).
-
Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica gel R6 as the coating substance and a mixture of 50 volumes of heptane R, 30 volumes of glacial acetic acid R and 20 volumes of dichloromethane R as the mobile phase. Apply separately to the plate, 5 μL of each of the following two solutions in ethanol R. For solution (A), use 10 mg of the test substance per mL and, for solution (B), use 2 mg of fumaric acid R per mL. Develop the plate in an unsaturated tank over a path of 10 cm. After removing the plate from the chromatographic chamber, allow it to dry exhaustively in air or in a current of air. Examine the chromatogram in ultraviolet light (254 nm).
One of the principal spots obtained with solution (A) corresponds in position, appearance and intensity with that obtained with solution (B).
Fumaric acid. Dissolve 0.250 g in 50 mL of water R and titrate with sodium hydroxide (0.1 mol/L) VS. Determine the end-point potentiometrically as described under 2.6 Non-aqueous titration, Method A. Each mL of sodium hydroxide (0.1 mol/L) VS is equivalent 5.804 mg of fumaric acid (C4H4O4). The fumaric acid content is between 17.5%-19.0% on the anhydrous basis.
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A. Use about 1.0 g of the substance; the water content is not more than 10 mg/g.
Heavy metals. Use 1.0 g in 30 mL of methanol R for the preparation of the test solution as described under 2.2.3 Limit test for heavy metals, Procedure 2; determine the heavy metals content according to Method A; not more than 20 μg/g.
Sulfated ash (2.3). Not more than 2.0 mg/g.
Impurity G (tenofovir disoproxil (S)-enantiomer). Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using a stainless steel column (15 cm x 4.6 mm) packed with particles of silica gel, the surface of which has been modified with amylose tris (3,5-dimethylphenyl carbamate) (5 µm). As the mobile phase, use a mixture of 900 volumes of dehydrated ethanol R, 100 volumes of acetonitrile R and 1 volume of diethylamine R.
Operate at a flow rate of 1.0 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 260 nm. Maintain the column temperature at 35 °C.
Prepare the following solutions using a mixture of 9 volumes of dehydrated ethanol R and 1 volume of acetonitrile (v/v) as the diluent. For solution (1), dissolve 40.0 mg of the test substance in 100.0 mL. For solution (2), dilute 1.0 mL of solution (1) to 100.0 mL. For solution (3), dilute 1.0 mL of solution (2) to 10.0 mL. For solution (4), dissolve the content of a vial of tenofovir disoproxil fumarate for enantiomer identification RS in 0.5 mL.
Inject 10 µL of solutions (3) and (4). Record the chromatograms for about 3 times the retention of tenofovir disoproxil.
Use the chromatogram obtained with solution (4), and the chromatogram supplied with tenofovir disoproxil fumarate for enantiomer identification RS, to identify the peak due to the impurity G. Impurity G is eluted at the relative retention of 1.6 with reference to tenofovir disoproxil (retention time about 3.5 minutes).
The test is not valid unless, in the chromatogram obtained with solution (4), the resolution factor between the peaks due to impurity G and tenofovir disoproxil is at least 5. Also, the test is not valid unless, in the chromatogram obtained with solution (3), the signal-to-noise ratio of the peak due to tenofovir disoproxil is at least 10.
Inject 10 µL each of solutions (1) and (2).
In the chromatogram obtained with solution (1):
-
the area of any peak corresponding to impurity G is not greater than the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (2) (1.0%).
Related substances. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using a stainless steel column (25 cm x 4.6 mm) packed with end-capped particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups (5 µm).
Use the following conditions for gradient elution:
- Mobile phase A: acetate buffer pH 4.2;
- Mobile phase B: acetonitrile R.
Prepare the acetate buffer pH 4.2 by dissolving 9.64 g of ammonium acetate R in 900 mL of water R, adjust the pH to 4.2 with glacial acetic acid R and dilute to 1000 mL with water R.
|
Time |
Mobile phase A |
Mobile phase B |
Comments |
|
0–2 |
100 |
0 |
Isocratic |
|
2–17 |
100 to 95 |
0 to 5 |
Linear gradient |
|
17–47 |
95 to 60 |
5 to 40 |
Linear gradient |
|
47–62 |
60 to 25 |
40 to 75 |
Linear gradient |
|
62–63 |
25 to 100 |
75 to 0 |
Return to initial composition |
|
63–75 |
100 |
0 |
Re-equilibration |
Operate with a flow rate of 1.0 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 260 nm. Maintain the column temperature at 25 °C and the autosampler temperature at 6 °C.
Prepare the following solutions using water R as diluent.
For solution (1), transfer 90 mg of test substance to a 100 mL volumetric flask. Add about 75 mL and sonicate at room temperature for about 30 minutes with intermittent shaking. Allow to cool to room temperature, dilute to volume and filter.
For solution (2), dilute 1.0 mL of solution (1) to 100.0 mL.
For solution (3), dilute 10.0 mL of solution (2) to 100.0 mL.
For solution (4), dissolve the content of a vial of tenofovir disoproxil fumarate for system suitability RS (containing tenofovir disoproxil fumarate and impurity H) in 0.5 mL.
For solution (5) (used to generate tenofovir disoproxil impurity A), dissolve 10 mg of tenofovir disoproxil fumarate RS in 10 mL of water R. Heat the solution carefully in a boiling water bath for 20 minutes.
For solution (6), use a solution containing 0.2 mg of fumaric acid R per mL of water R.
Inject 10 µL each of solutions (1), (2), (3), (4), (5) and (6).
Use the chromatogram obtained with solution (4) and the chromatogram supplied with tenofovir disoproxil fumarate for system suitability RS to identify the peak due to the tenofovir disoproxil impurity H in the chromatogram obtained with solution (1), if present.
Use the chromatogram obtained with solution (5) to identify the peak due to the tenofovir disoproxil impurity A in the chromatogram obtained with solution (1), if present.
Use the chromatogram obtained with solution (6) to identify the peak due to the fumaric acid in the chromatogram obtained with solution (1) if present. Fumaric acid is eluted at about 2.5 minutes and may appear as single or split peaks.
The impurities, if present, are eluted at the following relative retentions with reference to tenofovir disoproxil (retention time about 48 minutes):
|
Impurity |
Relative retention |
Impurity Classification |
|
Tenofovir disoproxil impurity R |
0.30 |
|
|
Tenofovir disoproxil impurity N |
0.33 |
Synthesis/Degradation |
|
Tenofovir disoproxil impurity A |
0.63 |
Synthesis/Degradation |
|
Tenofovir disoproxil impurity F |
0.73 |
Degradation |
|
Tenofovir disoproxil impurity E |
0.76 |
Synthesis/Degradation |
|
Tenofovir disoproxil impurity B |
0.80 and 0.81 |
Synthesis |
|
Tenofovir disoproxil impurity L |
0.87 |
Synthesis |
|
Tenofovir disoproxil impurity C |
0.88 |
Synthesis |
|
Tenofovir disoproxil impurity D |
0.90 |
Synthesis |
|
Tenofovir disoproxil impurity M |
0.94 |
Synthesis |
|
Tenofovir disoproxil impurity P |
0.96 |
Synthesis |
|
Tenofovir disoproxil impurity O |
0.97 |
Synthesis |
|
Tenofovir disoproxil impurity I |
0.98 |
Synthesis/Degradation |
|
Tenofovir disoproxil impurity H |
1.01 |
Synthesis |
|
Tenofovir disoproxil impurity Q |
1.10 |
Synthesis/Degradation |
|
Tenofovir disoproxil impurity J |
1.19 |
Synthesis/Degradation |
Note: Tenofovir disoproxil impurities B and C may appear as single or split peaks. If they appear as split peaks, use the sum of the two peaks in the calculation of the concentration. ("Synthesis" stands for synthesis related impurity; "Degradation" for degradation product.)
The test is not valid unless:
-
in the chromatogram obtained with solution (3), the signal-to-noise ratio of the peak due to tenofovir disoproxil is at least 20; and
-
in the chromatogram obtained with solution (4), the resolution between the peaks due to tenofovir disoproxil and tenofovir disoproxil impurity H is at least 1.2.
[Note from the Secretariat. It is intended to use peak-to-valley ratios in the verification of the system suitability once the International Chemical Reference Substance on tenofovir disoproxil fumarate for system suitability has been established.]
In the chromatogram obtained with solution (1):
-
the area of any peak corresponding to impurity A, when multiplied by a correction factor of 0.7, is not greater than the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (2) (1.0%);
-
the area of any peak corresponding to impurity D is not greater than 3 times the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (3) (0.3%);
-
the area of any peak corresponding to impurity N, when multiplied by a correction factor of 0.53, is not greater than 1.5 times the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (3) (0.15%);
-
the area of any peak corresponding to impurity R, when multiplied by a correction factor of 0.23, is not greater than 1.5 times the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (3) (0.15%);
-
the area of any peak corresponding to either impurities M, J, H or Q is not greater than 1.5 times the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (3) (0.15%);
-
the area of any other impurity peak is not greater than the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (3) (0.10%).
-
The sum of the areas of all impurity peaks, including the corrected areas of any peaks corresponding to impurities A, N and R, is not greater than 2.0 times the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (2) (2.0%). Disregard any peak with an area less than 0.5 times the area of the peak due to tenofovir disoproxil in the chromatogram obtained with solution (3) (0.05%) and any peak due to fumaric acid.
Assay. Dissolve 0.400 g in 30 mL of glacial acetic acid R1 and titrate with perchloric acid (0.1 mol/L) VS, and determine the end-point potentiometrically as described under 2.6 Non-aqueous titration, Method A. Each mL of perchloric acid (0.1 mol/L) VS is equivalent to 63.55 mg of tenofovir disoproxil fumarate (C19H30N5O10P, C4H4O4).
Impurities
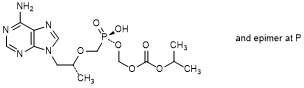
A. (Propan-2-yl) (8R)-9-(6-amino-9H-purin-9-yl)-5-hydroxy-8-methyl-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate (tenofovir monosoproxil), (synthesis-related impurity, degradation product).
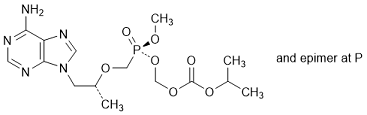
B. (Propan-2-yl) (5RS,8R)-9-(6-amino-9H-purin-9-yl)-5-methoxy-8-methyl-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate, (synthesis related impurity).
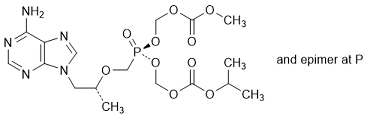
C. Methyl (propan-2-yl) (5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate, (synthesis-related impurity).
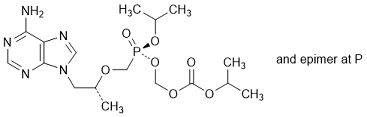
D. (Propan-2-yl) (5RS,8R)-9-(6-amino-9H-purin-9-yl)-8-methyl-5-(propan-2-yloxy)-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate, (synthesis-related impurity).
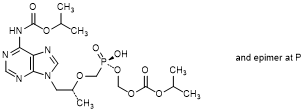
E. (Propan-2-yl) (8R)-5-hydroxy-8-methyl-9-(6-{[(propan-2-yloxy)carbonyl]amino}-9H-purin-9-yl)-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate, (synthesis-related impurity, degradation product).
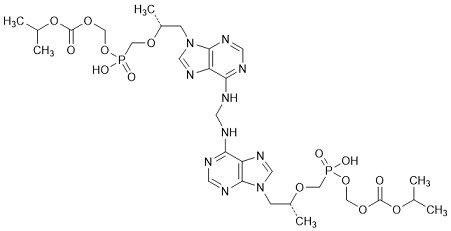
F. Bis(propan-2-yl) 9,9'-[methylenebis(imino-9H-purine-6,9-diyl)]bis[(8R)-5-hydroxy-8-methyl-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate] (tenofovir monosoproxil dimer), (degradation product).
[Note from the Secretariat: Structure will be added at a later stage.]
G. Bis(propan-2-yl) 5-{[(1S)-2-(6-amino-9H-purin-9-yl)-propan-2-yloxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate (tenofovir disoproxil (S)-enantiomer), (synthesis-related impurity).
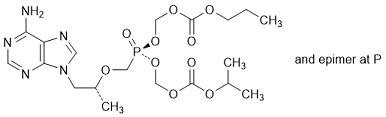
H. Propan-2-yl propyl (5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-propan-2-yloxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate (synthesis-related impurity).
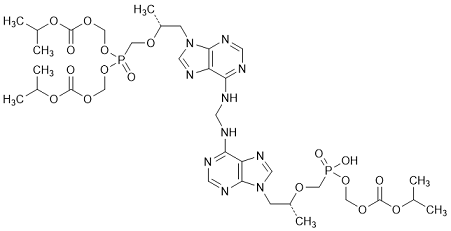
I. Bis(propan-2-yl) 5-{[(1R)-2-(6-{[({9-[(2R)-5-hydroxy-2,11-dimethyl-5,9-dioxo-3,6,8,10-tetraoxa-5-λ5-phosphadodecyl]-9H-purin-6-yl}amino)methyl]amino}-9H-purin-9-yl)-propan-2-yloxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate (tenofovir di- and monosoproxil heterodimer), (synthesis-related impurity, degradation product).
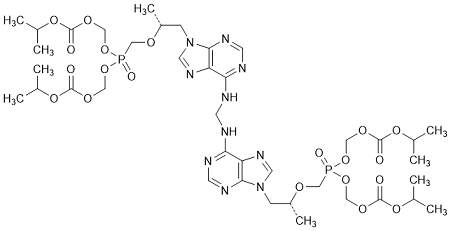
J. Tetrakis(propan-2-yl) 5,5'-(methylenebis{imino-9H-purine-6,9-diyl[(2R)-propane-1,2-diyl]oxymethylene})bis[5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate] (tenofovir disoproxil dimer), (synthesis-related impurity, degradation product).
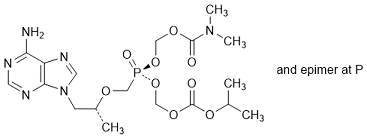
L. (Propan-2-yl) (5RS)-5-{[(2R)-1-(6-amino-9H-purin-9-yl)-propan-2-yloxy]methyl}-10-methyl-5,9-dioxo-2,4,6,8-tetraoxa-10-aza-5-λ5-phosphaundecanoate, (synthesis-related impurity)).
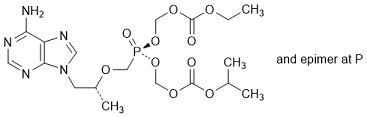
M. Ethyl propan-2-yl (5RS)-5-{[(1R)-2-(6-amino-9H-purin-9-yl)-propan-2-yloxy]methyl}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate, (synthesis-related impurity).
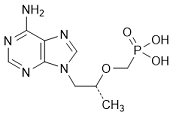
N. {[(1R)-1-(6-Amino-9H-purin-9-yl)-propan-2-yloxy]methyl}phosphonic acid (tenofovir), (synthesis-related impurity, degradation product).
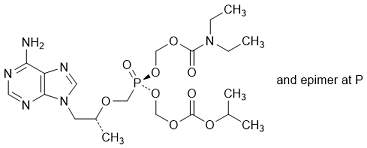
O. (Propan-2-yl) (5RS)-5-{[(2R)-1-(6-amino-9H-purin-9-yl)-propan-2-yloxy]methyl}-10-ethyl-5,9-dioxo-10-aza-2,4,6,8-tetraoxa-5-λ5-phosphadodecanoate (synthesis related impurity).
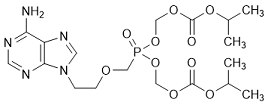
P. Bis(propan-2-yl) 5-{[2-(6-amino-9H-purin-9-yl)ethyl]oxymethyl]}-5-oxo-2,4,6,8-tetraoxa-5-λ5-phosphanonanedioate (desmethyl tenofovir disoproxil), (synthesis related impurity).
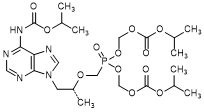
Q. Bis(propan-2-yl) (8R)-5-hydroxy-8-methyl-9-(6-{[(propan-2-yloxy)carbonyl]amino}-9H-purin-9-yl)-5-oxo-2,4,7-trioxa-5-λ5-phosphanonanoate, (synthesis-related impurity, degradation product).
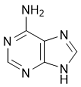
R. 6-Amino-9H-purine (adenine).