Monographs: Dosage forms: Specific monographs: Abacavir oral solution (Abacaviri solutio peroralum)
Category. Antiretroviral (Nucleoside Reverse Transcriptase Inhibitor).
Storage. Abacavir oral solution should be kept in a well-closed container.
Labelling. The designation of the container of Abacavir oral solution should state that the active ingredient is in the sulfate form and the quantity should be indicated in terms of the equivalent amount of abacavir.
Additional information. Strength in the current WHO Model list of essential medicines: 100 mg of abacavir per 5ml (20 mg per mL). Strength in the current WHO Model list of essential medicines for children: 100 mg of abacavir per 5ml (20 mg per mL).
Requirements
Complies with the monograph for "Liquid preparations for oral use".
Definition. Abacavir oral solution is a solution of Abacavir sulfate in a suitable vehicle, which may be flavoured. It contains not less than 90.0% and not more than 110.0% of the amount of abacavir (C14H18N6O) stated on the label.
Identity tests
• Either tests A, C and D or tests B, C and D may be applied.
A. Carry out test A.1 or, where UV detection is not available, test A.2.
A.1 Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica R6 as the coating substance and a mixture of 8 volumes of dichloromethane R, 2 volumes of 2-propanol R as the mobile phase. Apply separately to the plate 5 μl of each of the following 2 solutions. For solution (A) dilute a volume of the oral solution with methanol R to give a solution containing the equivalent of 5 mg of abacavir per mL. For solution (B) use 6 mg of abacavir sulfate RS per mL of methanol R. After removing the plate from the chromatographic chamber allow it to dry exhaustively in air or in a current of cool air. Examine the chromatogram in ultraviolet light (254 nm).
The principal spot obtained with solution A corresponds in position, appearance and intensity to that obtained with solution B.
A.2 Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using the conditions described above under test A.1 but using silica gel R5 as the coating substance. Spray with vanillin/sulfuric acid TS1. Heat the plate for a few minutes at 120 °C. Examine the chromatogram in daylight.
The principal spot obtained with solution (A) corresponds in position, appearance and intensity to that obtained with solution (B).
B. See the test described under Assay. The retention time of the principal peak in the chromatogram obtained with solution (1) is similar to that in the chromatogram obtained with solution (2).
C. To a volume of oral solution containing the equivalent of 15 mg of abacavir add 100 mL of water R and shake; dilute 5 mL of this solution to 50 mL with the same solvent. The absorption spectrum (1.6) of the resulting solution, when observed between 220 nm and 320 nm, exhibits a maximum at about 291 nm.
D. To a volume of the oral solution containing the equivalent of about 20 mg of abacavir add 5 mL of water R and shake. The resulting solution yields reaction A described under 2.1 General identification tests as characteristic of sulfates.
pH value (1.13). pH of the oral solution, 3.8–5.0.
Related substances
• Apply criteria A in all cases and criteria B whenever possible.
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using the same chromatographic conditions as described under Assay.
Prepare the following solutions in the dissolution solvent prepared by diluting 1 mL of phosphoric acid (~1440 g/l) TS in 1 litre of water R.
For solution (1) transfer a volume of the oral solution containing the equivalent of 10 mg of Abacavir in the dissolution solvent and dilute to 50.0 mL with the same solvent. For solution (2) dilute 5.0 mL of solution (1) to 50.0 mL with the dissolution solvent; dilute 5.0 mL of this solution to 50.0 mL with the same solvent. For solution (3) dissolve 5 mg of abacavir sulfate for system suitability RS (containing abacavir sulfate and impurities B to F) in the dissolution solvent and dilute to 25 mL with the same solvent. For solution (4) dissolve a suitable amount of each of the excipients stated on the label in 10 mL of a suitable solvent and dilute to 100 mL with the dissolution solvent.
Inject separately 20 μl each of solutions (1), (2), (3) and (4) and of dissolution solvent in the chromatographic system. Examine the blank chromatogram for any extraneous peaks and disregard the corresponding peaks observed in the chromatogram obtained with solution (1).
In the chromatogram obtained with solution (3) the impurity peaks are eluted at the following relative retention with reference to abacavir (retention time about 19 minutes): impurity C about 0.7; impurity D about 1.05; impurity E about 1.10; impurity B about 1.3; impurity F about 1.7. The test is not valid unless the resolution between the peaks due to abacavir and impurity D is at least 1.5.
If information concerning the excipients used in manufacturing the oral solution is not available or, if any of the peaks in the chromatogram obtained with solution (4) corresponds to any of the peaks in the chromatogram obtained with solution (3) or, if interference by excipients has been demonstrated by any other means, apply only criteria A.
A. In the chromatogram obtained with solution (1) the area of any peak corresponding to impurity C is not greater than the area of the principal peak in the chromatogram obtained with solution (2) (1.0%); the area of any peak with a relative retention greater than 0.5 but less than that of impurity C is not greater than 0.5 times the area of the principal peak in the chromatogram obtained with solution (2) (impurity G, 0.5%).
B. In the chromatogram obtained with solution (1) the area of any other peak, apart from the principal peak, is not greater than 0.3 times the area of the principal peak in the chromatogram obtained with solution (2) (0.3%). The sum of the areas of all peaks, other than the principal peak, is not greater than twice the area of the principal peak in the chromatogram obtained with solution (2) (2.0%). Disregard any peak with the same retention time as that of any of the peaks in the chromatogram obtained with solution (4) and any peak with an area less than 0.1 times the area of the principal peak in the chromatogram obtained with solution (2) (0.1%).
Assay
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using a stainless steel column (15 cm x 4.6 mm), packed with octadecylsilyl silica gel for chromatography (5 μm).
The mobile phases for gradient elution consist of a mixture of Mobile phase A and Mobile phase B, using the following conditions:
Mobile phase A: 0.05% solution of trifluoroacetic acid R in water R.
Mobile phase B: 85 volumes of methanol R and 15 volumes of water R.
| Time
(min) |
Mobile phase A
(% v/v) |
Mobile phase B
(% v/v) |
Comments |
| 0 - 20 | 95 to 70 | 5 to 30 | Linear gradient |
| 20 - 35 | 70 to 10 | 30 to 90 | Linear gradient |
| 35 - 40 | 10 | 90 | Isocratic |
| 40 - 41 | 10 to 0 | 90 to 100 | Linear gradient |
| 41 - 50 | 0 | 100 | Isocratic |
| 50 - 51 | 0 to 95 | 100 to 5 | Return to initial composition |
| 51 - 55 | 95 | 5 | Re-equilibration |
Operate with a flow rate of 0.8 mL per minute and the column oven temperature at 30 °C. As a detector use an ultraviolet spectrophotometer set at a wavelength of about 254 nm.
Prepare the following solutions in the dissolution solvent prepared by diluting 1 mL of phosphoric acid (~1440 g/l) TS in 1 litre of water R.
For solution (1) transfer an accurately weighed quantity of the oral solution containing the equivalent of about 10 mg of abacavir into a 50 mL volumetric flask. Add about 40 mL of dissolution solvent, shake mechanically for about 10 minutes and make up to volume using the same solvent. For solution (2) use 0.23 mg of abacavir sulfate RS per mL of dissolution solvent. For solution (3) dissolve 5 mg of abacavir sulfate for system suitability RS (containing abacavir sulfate and impurities B to F) in the dissolution solvent and dilute to 25 mL with the same solvent. For solution (4) dissolve a suitable amount of each of the excipients stated on the label in 10 mL of a suitable solvent and dilute to 100 mL with the dissolution solvent.
Inject separately 20 μl each of solutions (3) and (4). In the chromatogram obtained with solution (3) the impurity peaks are eluted at the following relative retention with reference to abacavir (retention time about 19 minutes): impurity C about 0.7; impurity D about 1.05; impurity E about 1.10; impurity B about 1.3; impurity F about 1.7. The assay is not valid unless the resolution between the peaks corresponding to abacavir and impurity D is at least 1.5. The assay is also not valid if any of the peaks in the chromatogram obtained with solution (4) corresponds to the peak due to abacavir in the chromatogram obtained with solution (3).
Inject alternatively 20 µl each of solution (1) and (2) and record the chromatograms. Measure the areas of the peak responses obtained in the chromatograms from solution (1). Determine the weight per mL (1.3.1) of the oral solution and calculate the percentage content of abacavir (C14H18N6O) weight in volume in the oral solution using the declared content of (C14H18N6O)2,H2SO4 in abacavir sulfate RS. Each mg of (C14H18N6O)2,H2SO4 is equivalent to 0.8537 mg of C14H18N6O.
Impurities
The impurities limited by the requirements of this monograph include those listed in the monograph for Abacavir sulfate and the following:
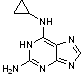
G. N6-cyclopropyl-1H-purine-2,6-diamine.