Monographs: Dosage forms: Specific monographs: Remdesivir intravenous infusion (Remdesiviri infusio intraveno)
Category. Antiviral.
Requirements
The intravenous infusion complies with the monograph on Parenteral preparations.
Definition. Remdesivir intravenous infusion is a sterile solution containing Remdesivir. It is prepared by reconstituting Remdesivir powder for concentrate for solution for infusion and diluting it with suitable diluents according to the manufacturer's instructions.
Remdesivir powder for concentrate for solution for infusion
Description. A white to off-white to slightly yellow powder or cake.
Storage. Remdesivir powder for concentrate for solution for infusion should be kept in sealed container, protected from moisture and light.
Manufacture. Remdesivir powder for concentrate for solution for infusion may contain solubilizing agents, like cyclodextrins. The production method is validated to ensure that the active pharmaceutical ingredient is fully dissolved before lyophilization.
Additional information. Strength available: 100 mg in vial.
Requirements
The powder for concentrate for solution for infusion complies with the monograph for Parenteral preparations.
Definition. Remdesivir powder for concentrate for solution for infusion is a sterile material consisting of Remdesivir with excipients. It is supplied in a sealed container and contains not less than 90.0% and not more than 110.0% of the labelled amount of Remdesivir (C27H35N6O8P) per vial.
Identity tests
- Either test A alone or any two of tests B, C and D may be applied.
-
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using the conditions given under "Assay", Method A but using, as the detector, a diode array detector to record the UV spectrum of the principal peak in each chromatogram in the range of 220 nm to 400 nm. The retention time and the UV spectrum of the principal peak in the chromatogram obtained with solution (1) correspond to the retention time and the UV spectrum of the peak due to remdesivir in the chromatogram obtained with solution (2).
-
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using the conditions given under "Assay", Method A. The retention time of the principal peak in the chromatogram obtained with solution (1) corresponds to the retention time of the peak due to remdesivir in the chromatogram obtained with solution (2).
-
Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica gel R6 as the coating substance and a freshly prepared mixture of ethyl acetate R, methanol R and glacial acetic acid R (90:9:1 V/V/V) as the mobile phase. Apply separately to the plate 2 µL of each of the following two solutions. For solution (A), add 20 mL of methanol R to a vial and re-insert the stopper. Swirl the vial content gently and then mix well by repeated inversions. Transfer the dispersion into a 100 mL volumetric flask and add 60 mL of methanol R. Sonicate the flask for 15 minutes, dilute to volume with methanol R and filter the dispersion and use the filtrate. For solution (B), use a solution containing 1 mg of remdesivir RS per mL of methanol. After removing the plate from the chromatographic chamber allow it to dry in air or in a current of air. Examine the chromatogram under ultraviolet light (254 nm). The principal spot in the chromatogram obtained with solution (A) corresponds in position, appearance and intensity with the spot due to remdesivir in the chromatogram obtained with solution (B).
-
Prepare the test solution as described under "Assay", Method B. The absorption spectrum (1.6 Spectrophotometry in the visible and ultraviolet regions) of the solution, when observed between 220 nm and 400 nm, exhibits two maxima at about 245 nm and 275 nm.
pH (1.13). Reconstitute Remdesivir powder for concentrate for solution for infusion according to the manufacturer's instructions, but using carbon-dioxide free water R as the solvent. pH of the reconstituted solution, 3.0 to 4.0.
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A. Use 0.200 g of the test substance. The water content is not more than 30 mg/g.
Related substances. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using a stainless steel column (4.6 mm x 15 cm) packed with particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups (3 µm).
Use the following conditions for gradient elution:
- Mobile phase A: phosphoric acid solution;
- Mobile phase B: acetonitrile for chromatography R.
Prepare the phosphoric acid solution by diluting 1.2 mL of phosphoric acid (~1440 g/L) TS to 1000 mL with water R.
|
Time (minutes) |
Mobile phase A (% v/v) |
Mobile phase B (% v/v) |
Comments |
|
0–3 |
97 |
3 |
Isocratic |
|
3–45 |
97 to 30 |
3 to 70 |
Linear gradient |
|
45–53 |
30 |
70 |
Isocratic |
|
53–54 |
30 to 97 |
70 to 3 |
Return to initial composition |
|
54–65 |
97 |
3 |
Re-equilibration |
Operate with a flow rate of 1.0 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 237 nm. Maintain the column temperature at 30 °C.
Prepare as a diluent a mixture of 50 volumes of methanol R and 50 volumes of water R.
Prepare the following solutions. For solution (1), gently remove the stopper from a vial and retain the stopper. Add 20 mL of water R to the vial and re-insert the stopper. Swirl the vial content gently and then mix well by repeated inversions until the content of the vial is fully dissolved. Transfer the solution into a 200 mL volumetric flask and dilute to volume with the diluent. For solution (2), dilute 1.0 mL of solution (1) to 100.0 mL with the diluent. For solution (3), dilute 1.0 mL of solution (2) to 10.0 mL with the diluent. For solution (4), dissolve and dilute remdesivir for system suitability RS (containing remdesivir and impurity A) as described in the leaflet of the reference substance.
Inject 5 µL each of solutions (1), (2), (3) and (4).
Use the chromatogram obtained with solution (4) and the chromatogram supplied with remdesivir for system suitability RS to identify the peak due to impurity A. The impurities are eluted, if present, at the following relative retentions with reference to remdesivir (retention time about 28 minutes): impurity E about 0.18; impurity G about 0.46; impurity H about 0.51; impurity I about 0.74; impurity C and impurity D about 0.84; impurity A about 0.98; impurity B about 1.03 and impurity F about 1.30.
The test is not valid unless, in the chromatogram obtained with solution (4), the resolution between the peaks due to impurity A and remdesivir is at least 1.5. Also, the test is not valid unless in the chromatogram obtained with solution (3), the peak due to remdesivir is obtained with a signal-to-noise ratio of at least 10.
In the chromatogram obtained with solution (1):
-
the area of any peak corresponding to impurities A, F, G or I is not greater than 0.5 times the area of the peak due to remdesivir in the chromatogram obtained with solution (2) (0.5 %);
-
the area of any peak corresponding to impurity E, when multiplied with a correction factor of 0.5, is not greater than twice the area of the peak due to remdesivir in the chromatogram obtained with solution (2) (2.0 %);
-
the area of any other impurity peak is not greater than 2 times the area of the peak due to remdesivir in the chromatogram obtained with solution (3) (0.2 %).
-
The sum of the areas of all impurity peaks, including the corrected area of any peak corresponding to impurities E, is not greater than three times the area of the peak due to remdesivir in the chromatogram obtained with solution (2) (3.0%). Disregard all peaks with an area of less than the area of the peak due to remdesivir in the chromatogram obtained with solution (3) (0.1%).
Assay
- Either test A or test B may be applied.
-
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using a stainless steel column (4.6 mm x 25 cm) packed with end-capped particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups (5 µm).
Prepare a phosphoric acid solution by diluting 1.2 mL of phosphoric acid (~1440 g/L) TS to 1000 mL with water R. As the mobile phase, use a mixture of 35 volumes of the phosphoric acid solution and 65 volumes of methanol R.
Operate with a flow rate of 1.0 mL per minute.
As a detector, use an ultraviolet spectrophotometer set at a wavelength of 237 nm. Maintain the column temperature at 25 °C.
Prepare as a diluent a mixture of 50 volumes of methanol R and 50 volumes of water R.
Prepare the following solutions. For solution (1), gently remove the stoppers from 10 vials and retain the stoppers. Add 20 mL of water R to each vial and re-insert the stoppers. Swirl the vial contents gently and then mix well by repeated inversions until the contents of the vials are fully dissolved. Transfer the solutions into a single 500 mL volumetric flask. Add 10 mL of water R to each vial, re-insert the stoppers, mix and transfer the rinses to the volumetric flask. Dilute to volume with water R. Dilute 5.0 mL of this solution to 50.0 mL with the diluent.
For solution (2), weigh 40.0 mg of remdesivir RS into a 200 mL volumetric flask. Dissolve with 10 mL of methanol R and make up to volume with the diluent.
Inject 5 µL each of solutions (1) and (2) and record the chromatogram for 25 minutes.
Measure the areas of the peaks corresponding to remdesivir obtained in the chromatograms of solutions (1) and (2) and calculate the percentage content of C27H35N6O8P per container, using the declared content of C27H35N6O8P in remdesivir RS.
-
Prepare solution (1) as described under "Assay", Method A. Dilute 5.0 mL of this solution to 20.0 mL with a mixture of 50 volumes of methanol R and 50 volumes of water R. Measure the absorbance of the resulting test solution as described under 1.6 Spectrophotometry in the visible and ultraviolet regions in a cuvette with an optical pathlength of 10 mm at the maximum at about 275 nm, using the diluent as the blank.
Calculate the percentage content of C27H35N6O8P per container using the absorptivity value of 9.984 for remdesivir
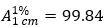 .
.
Bacterial endotoxins. Carry out the test as described under 3.4 Test for bacterial endotoxins; contains not more than 1.0 IU of endotoxin RS per mg of Remdesivir.
Impurities. The impurities limited by the requirements of this monograph include those listed in the monograph on Remdesivir.