Monographs: Dosage forms: Specific monographs: Sofosbuvir tablets (Sofosbuvir compressi)
Category. Antiviral (Hepatitis C viral polymerase nucleotide inhibitor).
Storage. Sofosbuvir tablets should be kept in a well-closed container not exceeding 30 °C.
Additional information. Strength in the current WHO Model List of Essential Medicines: 400 mg sofosbuvir.
Requirements
Comply with the monograph for Tablets.
Definition. Sofosbuvir tablets contain not less than 90.0 % and not more than 110.0 % of the amount of sofosbuvir (C22H29FN3O9P) stated on the label.
Identity tests
-
Either test A alone or tests B and D or tests C and D may be applied.
-
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions given under "Assay". Record the UV spectrum of the principal peak in the chromatograms with a diode array detector in the range of 200 nm to 400 nm. The retention time and the UV spectrum of the principal peak in the chromatogram obtained with solution (1) correspond to the retention time and UV spectrum of the peak due to sofosbuvir in the chromatogram obtained with solution (2).
-
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions given under "Assay". The retention time of the principal peak in the chromatogram obtained with solution (1) corresponds to the retention time of the peak due to sofosbuvir in the chromatogram obtained with solution (2).
-
Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica gel R2 as the coating substance and a mixture of 6 volumes of dichloromethane R, 1 volume of methanol R, 4 volumes of ethyl acetate R and 0.1 volume of ammonia (~260 g/L) TS as the mobile phase.
Apply separately to the plate 10 µL of each of the following solutions in methanol R. For solution (A), sonicate a quantity of the powdered tablets, nominally containing 10 mg of sofosbuvir with 10 mL of methanol R, allow to cool to room temperature and filter. For solution (B), use 1 mg of sofosbuvir RS per mL. After removing the plate from the chromatographic chamber, allow it to dry exhaustively in a current of air. Examine the chromatogram in ultraviolet light (254 nm). The principal spot obtained with solution (A) corresponds in position, appearance and intensity to that obtained with solution (B).
-
To a quantity of the powdered tablets, nominally containing 50 mg of sofosbuvir, add 30 mL of methanol R and sonicate for 10 minutes. Allow to cool to room temperature, dilute to 50 mL with the same solvent and filter. Dilute 1.0 mL of the filtrate to 100.0 mL using methanol R. The absorption spectrum (1.6) of the resulting solution, when observed between 200 and 400 nm, exhibits a maximum at about 260 nm.
Dissolution. Carry out the test as described under 5.5 Dissolution test for solid oral dosage forms using as dissolution medium 900 mL of dissolution buffer pH 6.8 TS. Rotate the paddle at 75 revolutions per minute. At 15 minutes, withdraw a sample of 10 mL of the medium through an in-line filter. Allow the filtered solution to cool down to room temperature. Dilute 5.0 mL of the filtrate to 50.0 mL with dissolution medium and use it as solution (1). For solution (2), transfer 44.0 mg of sofosbuvir RS to a 100 mL volumetric flask. Add 4 mL of methanol R and 70 mL of dissolution medium, sonicate for 5 minutes, cool to room temperature and dilute to volume. Dilute 5.0 mL of this solution to 50.0 mL with dissolution medium.
Measure the absorbance as described under 1.6 Spectrophotometry in the visible and ultraviolet regions of a 1 cm layer of the resulting solutions at the maximum at about 262 nm, using the dissolution medium as the blank.
For each of the tablets tested, calculate the total amount of sofosbuvir (C22H29FN3O9P) in the medium. Evaluate the results as described under 5.5 Dissolution test for solid oral dosage forms , Acceptance criteria. The amount of sofosbuvir released is not less than 75% (Q) of the amount declared on the label.
[Note from the Secretariat. It is intended to determine the absorptivity value of sofosbuvir during the establishment of the corresponding International Chemical Reference Standard and to use this value for the calculation of the test result.]
Related substances. Carry out the test as described under 1.14.4 High–performance liquid chromatography, using a column (150 mm x 4.6 mm) packed with end-capped, base deactivated particles of silica gel, the surface of which has been modified with chemically bonded octadecylsilyl groups (3.5 μm). Use the following conditions for gradient elution:
As mobile phase A, use a mixture of 21 volumes of 0.05 % phosphoric acid (~1440 g/L) TS, 77 volumes of water R and 2 volumes of acetonitrile R. As mobile phase B, use a mixture of 21 volumes of 0.05 % phosphoric acid (~1440 g/L) TS and 79 volumes of acetonitrile R.
Use the following gradient:
|
Time (min) |
Mobile phase A (% v/v) |
Mobile phase B (% v/v) |
Comments |
|
0 - 2.5 |
100 |
0 |
Isocratic |
|
2.5 - 27.4 |
100 to 0 |
0 to 100 |
Linear gradient |
|
27.4 - 38 |
0 |
100 |
Isocratic |
|
38 - 45 |
100 |
0 |
Re-equilibration |
Operate with a flow rate of 1.5 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 260 nm. Maintain the column temperature at 30 °C.
Prepare the following solutions using as diluent a mixture of 50 volumes of mobile phase A and 50 volumes of mobile phase B. Prepare solution (1) as described under "Assay". For solution (2), dilute 1.0 mL of solution (1) to 100.0 mL. For solution (3), dilute 5.0 mL of solution (2) to 50.0 mL. For solution (4), dissolve 1.0 mg of sofosbuvir for peak identification RS (containing sofosbuvir and the impurities A, B, C, D and E) in 5.0 mL. For solution (5), dissolve 25.0 mg of phenol R and dilute to 50.0 mL. Dilute 2.0 mL to of this solution to 100.0 mL. Dilute 5.0 mL of this solution to 50.0 mL.
Inject 20 µL of solutions (3) and (4).
The test is not valid unless the peak-to-valley ratio (Hp/Hv) in the chromatogram obtained with solution (4) is at least x[1], where Hp is the height above the extrapolated baseline of the peak due to impurity A and Hv is the height above the extrapolated baseline at the lowest point of the curve separating this impurity from the peak due to sofosbuvir. Also, the test is not valid unless the signal-to-noise of the peak due to sofosbuvir in the chromatogram obtained with solution (3) is at least 10.
[1] A value for the Hp/Hv has to be determined based on the available sofosbuvir for peak identification RS.
Inject alternately 20 µL each of solutions (1), (2) and (5).
Use the chromatograms obtained with solutions (4) and (5) and the relative retentions below to identify the peaks due to impurities D, 1, 2, 3, 4 and 5.
The impurities, if present, are eluted at the following relative retentions with reference to sofosbuvir (retention time about 16 minutes): impurity A about 0.98; impurity B about 1.05; impurity C about 1.41; impurity D about 0.38; impurity E about 0.93, impurity F about 1.13; impurity G about 1.57; impurity 1 about 0.21; impurity 2 about 0.45; impurity 3 about 0.61; impurity 4 about 0.65; impurity 5 about 0.72.
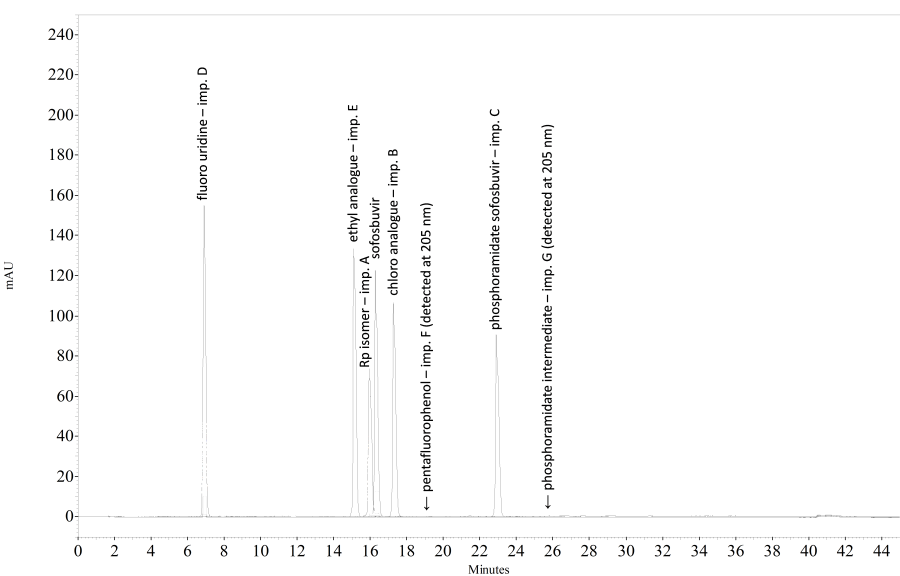
[Note from the Secretariat. The chromatogram shows, by way of example, the separation of sofosbuvir and its impurities.]
In the chromatogram obtained with solution (1):
-
the area of any peak corresponding to impurity D, when multiplied by a correction factor of 0.5, is not greater than 0.2 times the area of the peak due to sofosbuvir in the chromatogram obtained with solution (2) (0.2 %);
-
the area of any peak corresponding to impurities 1, 2, 3 or 4 is not greater than 0.2 times the area of the peak due to sofosbuvir in the chromatogram obtained with solution (2) (0.2 %);
-
the area of any peak corresponding to impurity 5 is not greater than the area of the peak due to phenol in the chromatogram obtained with solution (5) (0.2 %).
-
The sum of the areas of all impurity peaks, including the corrected area of any peak corresponding to impurity D, is not greater than the area of the peak due to sofosbuvir in the chromatogram obtained with solution (2) (1.0 %). Disregard any peak with an area less than the area of the peak due to sofosbuvir in the chromatogram obtained with solution (3) (0.1%).
Assay. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions given under "Related substances" with the following modifications:
As the mobile phase use a mixture of 65 volumes of mobile phase A and 35 volumes of mobile phase B.
Prepare the following solutions using the mobile phase as diluent. For solution (1), weigh and powder 20 tablets. Transfer a quantity of the powdered tablets, nominally containing 125.0 mg of sofosbuvir, to a 250 mL volumetric flask. Add about 200 mL of diluent and sonicate for 10 minutes, cool to room temperature, dilute to volume and filter. For solution (2), dilute 50.0 mg of sofosbuvir RS and dilute to 100.0 mL.
Inject alternatively 20 µL each of solutions (1) and (2). Record the chromatograms for 20 minutes. The retention time of sofosbuvir is about 11 minutes.
Measure the areas of the peaks corresponding to sofosbuvir obtained in the chromatograms from solutions (1) and (2) and calculate the percentage content of C22H29FN3O9P, using the declared content of C22H29FN3O9P in sofosbuvir RS.
Impurities. The impurities limited by the requirements of this monograph include the impurities listed in the monograph for sofosbuvir and the impurities 1, 2, 3, 4 and 5.
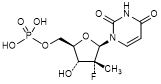
1. (2'R)-2'-Deoxy-2'-fluoro-2'-methyluridine 5'-(dihydrogen phosphate) (fluoro uridine phosphate) (degradation product).
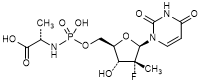
2. N-[{[(2R,3R,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy}(hydroxy)phosphoryl]-l-alanine (uridine alanine phosphate) (degradation product),
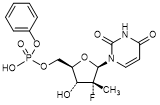
3. (2'R)-2'-Deoxy-2'-fluoro-5'-O-[hydroxy(phenoxy)phosphoryl)-2'-methyluridine (uridine phenyl phosphate) (degradation product),
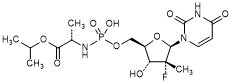
4. Propan-2-yl N-[{[(2R,3R,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy}(hydroxy)phosphoryl]-l-alaninate (uridine isopropyl alanine phosphate) (degradation product),
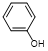
5. Phenol (degradation product).