Monographs: Pharmaceutical substances: Moxifloxacin hydrochloride (Moxifloxacini hydrochloridum)
Molecular formula. C21H25ClFN3O4, H2O
Relative molecular mass. 455.9
Graphic formula.
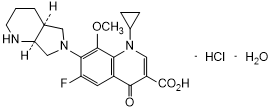
Chemical name. 1-Cyclopropyl-6-fluoro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo [3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid monohydrochloride monohydrate (IUPAC);1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid, hydrochloride, hydrate (1:1:1) (CAS); CAS Reg. No. 192927-63-2.
Description. A light yellow or yellow powder or crystals.
Solubility. Sparingly soluble in water R, slightly soluble in ethanol (~ 760 g/L) TS, practically insoluble in acetone R.
Category. Antibacterial, antituberculosis.
Storage. Moxifloxacin hydrochloride should be kept in tightly closed containers, protected from light.
Labelling. The designation on the container of Moxifloxacin hydrochloride should state the substance is in the form of the monohydrate.
Additional information. Moxifloxacin hydrochloride may exhibit polymorphism.
Requirements
Definition. Moxifloxacin hydrochloride contains not less than 98.0% and not more than 102.0% (“Assay”, method A) or not less than 99.0% and not more than 101.0% (“Assay”, method B) of C21H25ClFN3O4, calculated with reference to the anhydrous substance.
Identity tests.
- Either tests A, D and E or tests B, C, D and E may be applied.
A. Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from moxifloxacin hydrochloride RS or the reference spectrum of moxifloxacin hydrochloride.
If the spectra thus obtained are not concordant, repeat the test using the residues obtained by separately dissolving the test substance and moxifloxacin hydrochloride RS in a small amount of methanol R and evaporating to dryness. The infrared absorption spectrum is concordant with the spectrum obtained from moxifloxacin hydrochloride RS.
B. Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography using silica gel R5 as the coating substance and a mixture of 4 volumes of 1-butanol R, 4 volumes of methanol R and 2 volumes of ammonia (~100 g/L) TS as the mobile phase. Apply separately to the plate 10 µL of each of the following two solutions: for solution (A), use a 0.05 mg/mL solution of the test substance in methanol R. For solution (B), use a 0.05 mg/mL solution of moxifloxacin hydrochloride RS in methanol R. Develop the plate for a distance of 15 cm. After removing the plate from the chromatographic chamber, allow it to dry in air or in a current of air. Examine the chromatogram under ultraviolet light (365 nm).
The principal spot in the chromatogram obtained with solution (A) corresponds in position, appearance and intensity with the spot due to moxifloxacin in the chromatogram obtained with solution (B).
C. Dissolve 25 mg of the test substance in 20 mL of methanol and dilute to 50.0 mL with the same solvent. Dilute 1.0 mL of this solution to 100.0 mL using methanol R. The absorption spectrum (1.6) of the resulting solution, when observed between 250 and 320 nm, exhibits a maximum at about 295 nm.
D. Carry out test D.1 or test D.2.
D.1 Determine the specific optical rotation (1.4) using a solution of 0.200 g of the test substance in 20.0 mL of a mixture of equal volumes of acetonitrile R and water R and calculate with reference to the anhydrous substance
= -125 to -138.
D.2 Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions and solutions given under “Impurity G (moxifloxacin enantiomer).” The retention time of the principal peak obtained with solution (1) corresponds to the retention time of the peak due to moxifloxacin in the chromatogram obtained with solution (3).
E. Dissolve 50 mg of the test substance in 5 mL of water R, add 1 mL of nitric acid (~130 g/L) TS, mix, allow to stand for 5 minutes and filter. The filtrate yields reaction A described under 2.1 General identification tests as characteristic of chlorides.
Clarity and colour of solution. Dissolve 1.0 g of the test substance in 20 mL of sodium hydroxide (~85 g/L) TS. The solution is not more opalescent than opalescence standard TS3 and not more intensely coloured than reference solution GY2 (1.11.2, Method II).
pH value (1.13). pH of a 2 mg/mL solution in carbon-dioxide-free water R, 3.9 to 4.6.
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A, using 0.200 g of the substance, 40 mL anhydrous methanol, and 3 minutes stirring before titration starts; the water content is not less than 30 mg/g and not more than 45 mg/g.
Sulfated ash (2.3). Not more than 1.0 mg/g, determined on 1.000 g in a platinum crucible.
Impurity G (moxifloxacin enantiomer). Perform the test in subdued light, preferably using low-actinic glassware.
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (15 cm x 3.0 mm), packed with end-capped, base-deactivated particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups (3 μm).
Prepare the following chiral reagent solution. Dissolve 0.47 g of anhydrous copper (II) sulfate R and 1.31 g of isoleucine R in 1000 mL of water R and adjust with sodium hydroxide (~4 g/L) TS to a pH of 4.50.
Use the following conditions for gradient elution:
- Mobile phase A: 500 volumes of methanol R and 1500 volumes of chiral reagent solution.
- Mobile phase B: 225 volumes of methanol R and 450 volumes of chiral reagent solution.
|
Time (minutes) |
Mobile phase A (% v/v) |
Mobile phase B (% v/v) |
Comments |
|
0–50 |
100 |
0 |
Isocratic |
|
50–51 |
100 to 0 |
0 to 100 |
Linear gradient |
|
51–61 |
0 |
100 |
Isocratic |
|
61–62 |
0 to 100 |
100 to 0 |
Return to initial composition |
|
62–85 |
100 |
0 |
Re-equilibration |
Operate with a flow rate of 0.42 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 295 nm. Maintain the column temperature at 25 °C.
Prepare the following solutions in mobile phase A. For solution (1), dissolve 5.0 mg of the test substance in 5.0 mL. For solution (2), dilute 3.0 mL of solution (1) to 200.0 mL. Dilute 1.0 mL of this solution to 10.0 mL. For solution (3), dissolve 5 mg of moxifloxacin for system suitability RS (containing moxifloxacin and impurity G) in 5.0 mL.
Inject alternately 1.5 µL of solution (1), (2) and (3).
In the chromatogram obtained with solution (3), the peak due to impurity G is eluted with a relative retention of about 0.78 with respect to the peak due to moxifloxacin (retention time about 35 minutes).
The test is not valid unless, in the chromatogram obtained with solution (3), the resolution between the peak due to impurity G and the peak due to moxifloxacin is at least 2.0. Also, the test is not valid unless in the chromatogram obtained with solution (2) the peak due to moxifloxacin is detected with a signal-to-noise ratio of at least 10.
In the chromatogram obtained with solution (1):
- the area of any peak corresponding to impurity G (moxifloxacin enantiomer) is not greater than the area of the peak due to moxifloxacin in the chromatogram obtained with solution (2) (0.15%).
Related substances. Perform the test in subdued light, preferably using low-actinic glassware.
Carry out the tests as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (25 cm × 4.6 mm) packed with end-capped, base-deactivated particles of silica gel, the surface of which has been modified with chemically-bonded phenyl groups (5 µm).
Use the following mobile phase: mix 28 volumes of methanol R and 72 volumes of a solution containing 0.5 g/L of tetrabutylammonium hydrogen sulfate R, 1.0 g/L of potassium dihydrogen phosphate R and 3.4 g/L of phosphoric acid (~1440 g/L) TS.
Operate with a flow rate of 1.3 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 293 nm. Maintain the column temperature at 45 °C.
Prepare solvent (A) by dissolving 0.50 g of tetrabutylammonium hydrogen sulfate R and 1.0 g of potassium dihydrogen phosphate R in about 500 mL of water R. Add 2 mL of phosphoric acid (~1440 g/L) TS and 0.050 g of anhydrous sodium sulfite R, then dilute to 1000.0 mL with water R.
Prepare the following solutions in solvent (A). For solution (1), dissolve 50.0 mg of the test substance and dilute to 50.0 mL. For solution (2), dilute 1.0 mL of solution (1) to 100.0 mL. Dilute 1.0 mL of this solution to 10.0 mL. For solution (3), dissolve 5 mg of moxifloxacin for peak identification A RS (containing moxifloxacin and the impurities A, B and E) in 5 mL. For solution (4), dissolve 2 mg of moxifloxacin for peak identification B RS (containing moxifloxacin and the impurity F) in 2 mL.
Inject alternately 10 µL of solutions (1), (2), (3) and (4). Record the chromatograms for 2.5 times the retention time of moxifloxacin.
Use the chromatogram supplied with moxifloxacin for peak identification A RS and the chromatogram obtained with solution (3) to identify the peaks due to impurities A, Band E. Use the chromatogram supplied with moxifloxacin for peak identification B RS and the chromatogram obtained with solution (4) to identify the peak due to impurity F. The impurities are eluted at the following relative retention with reference to moxifloxacin (retention time about 11 minutes): impurity F about 0.9, impurity A about 1.1; impurity B about 1.3 and impurity E about 1.7.
The test is not valid unless, in the chromatogram obtained with solution (3), the resolution between the peak due to moxifloxacin and the peak due to impurity A is at least 1.5. Also, the test is not valid unless, in the chromatogram obtained with solution (2), the peak due to moxifloxacin is detected with a signal-to-noise ratio of at least 40.
In the chromatogram obtained with solution (1):
- the area of any peak corresponding to impurity B, when multiplied by a correction factor of 1.4, is not greater than 1.5 times the area of the peak due to moxifloxacin in the chromatogram obtained with solution (2) (0.15%);
- the area of any peak corresponding to impurity E, when multiplied by a correction factor of 3.5, is not greater than 1.5 times the area of the peak due to moxifloxacin in the chromatogram obtained with solution (2) (0.15%);
- the area of any peak corresponding to impurity F is not greater than 1.5 times the area of the peak due to moxifloxacin in the chromatogram obtained with solution (2) (0.15%);
- the area of any other impurity peak is not greater than the area of the peak due to moxifloxacin in the chromatogram obtained with solution (2) (0.10%).
- The sum of the corrected areas of any peaks corresponding to impurities B and E and the areas of all other impurity peaks is not greater than 3 times the area of the peak due to moxifloxacin obtained with solution (2) (0.3%). Disregard any peak with an area or, in the case of impurities B and E, a corrected area of less than 0.5 times the area of the peak due to moxifloxacin in the chromatogram obtained with solution (2) (0.05%).
Assay.
- Either test A or test B may be applied.
A. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions given under “Related substances”.
Prepare the following solutions in solvent (A). For solution (1), dissolve 50.0 mg of the test substance to be examined and dilute to 50.0 mL. Dilute 2.0 mL of this solution to 20.0 mL. For solution (2), dissolve 50.0 mg of moxifloxacin hydrochloride RS and dilute to 50.0 mL. Dilute 2.0 mL of this solution to 20.0 mL.
Inject alternately 10 µL of solutions (1) and (2).
Measure the areas of the peaks corresponding to moxifloxacin obtained in the chromatograms of solutions (1) and (2) and calculate the percentage content of moxifloxacin hydrochloride (C21H25ClFN3O4) using the declared content of moxifloxacin hydrochloride (C21H25ClFN3O4) in moxifloxacin hydrochloride RS.
B. Dissolve 0.320 g of the test substance in 50 mL of water R. Titrate with sodium hydroxide (0.1 mol/L) VS, determining the end-point potentiometrically. Read the volume added to reach the first point of inflection. Each mL of sodium hydroxide (0.1 mol/L) VS is equivalent to 43.79 mg of C21H25ClFN3O4.
Impurities.
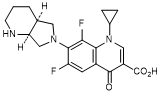
A. 1-Cyclopropyl-6,8-difluoro-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (synthesis-related impurity).
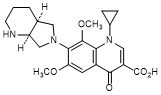
B. 1-Cyclopropyl-6,8-dimethoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (synthesis-related impurity).
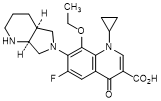
C. 1-Cyclopropyl-8-ethoxy-6-fluoro-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (synthesis-related impurity).
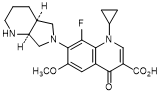
D. 1-Cyclopropyl-8-fluoro-6-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (synthesis-related impurity).
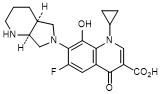
E. 1-Cyclopropyl-6-fluoro-8-hydroxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (synthesis-related impurity).
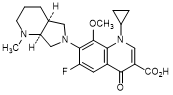
F. 1-Cyclopropyl-6-fluoro-8-methoxy-7-[(4aS,7aS)-1-methyloctahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (synthesis-related impurity).
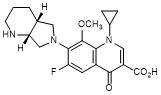
G. 1-Cyclopropyl-6-fluoro-8-methoxy-7-[(4aR,7aR)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (moxifloxacin enantiomer) (synthesis-related impurity).
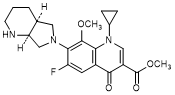
H. Methyl 1-cyclopropyl-6-fluoro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylate (synthesis-related impurity).