Monographs: Pharmaceutical substances: Dexamethasone sodium phosphate (Dexamethasone natrii phosphas)
Graphic formula
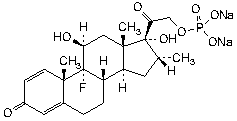
Molecular formula. C22H28FNa2O8P
Relative molecular mass. 516.4
Chemical name. 9-fluoro-11β,17-dihydroxy-16α-methyl-3,20-dioxopregna-1,4-dien-21-yl disodium phosphate; (11β,16α)-9-fluoro-11,17-dihydroxy-16-methyl-21-(phosphonooxy)pregna-1,4-diene-3,20-dione sodium salt (1:2); CAS Reg. No. 2392-39-4.
Description. A white, or almost white, crystalline powder.
Solubility. Freely soluble in water; slightly soluble in ethanol (~750 g/L) TS; practically insoluble in dichloromethane R.
Category. Adrenal hormone.
Storage. Dexamethasone sodium phosphate should be kept in a tightly closed container, protected from light.
Additional information. Dexamethasone sodium phosphate is very hygroscopic. Even in the absence of light, it is gradually degraded on exposure to a humid atmosphere, the decomposition being faster at higher temperatures. Dexamethasone sodium phosphate may exhibit polymorphism.
Requirements
Definition. Dexamethasone sodium phosphate contains not less than 97.0% and not more than 102.0% ("Assay", method A) or not less than 98.0% and not more than 102.0% ("Assay", method B) of C22H28FNa2O8P, calculated with reference to the anhydrous and ethanol-free substance.
Identity tests
-
Either tests A and F or tests B, D, E and F or tests C, D, E and F should be applied.
-
Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from dexamethasone sodium phosphate RS or with the reference spectrum of dexamethasone sodium phosphate.
-
Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography using silica gel R2 as the coating substance and a freshly prepared mixture of 60 volumes of 1-butanol R, 20 volumes of acetic acid (~300 g/L) TS and 20 volumes of water as the mobile phase. Apply separately to the plate 5 µL of each of the following 3 solutions in methanol R containing (A) 1 mg of the test substance per mL, (B) 1 mg of dexamethasone sodium phosphate RS per mL and (C) equal volumes of solution (A) and a solution of 1 mg of prednisolone sodium phosphate RS per mL of methanol R. After removing the plate from the chromatographic chamber, allow it to dry in air or in a current of air, spray it with sulfuric acid/ethanol (20%) TS, heat it at 120 °C for 10 minutes or until the spots appear, allow it to cool and examine the chromatogram in daylight and in ultraviolet light (365 nm). The principal spot obtained with solution (A) corresponds in position, appearance and intensity with that obtained with solution (B). The chromatogram of solution (C) shows 2 spots which may not, however, be completely separated.
-
See the test described under "Assay", method A. The retention time of the principal peak in the chromatogram obtained from solution (1) is similar to that in the chromatogram obtained from solution (2).
-
Dissolve 10.0 mg of the test substance in 5 mL of water R and dilute to 100.0 mL with dehydrated ethanol R. Transfer 2.0 mL of this solution to a glass-stoppered tube, add 10.0 mL of phenylhydrazine/sulfuric acid TS, mix and heat in a water bath at 60 °C for 20 minutes. Cool immediately. The absorbance (1.6) measured at the absorption maximum at about 419 nm is at least 0.20.
-
Heat carefully 0.04 g of the test substance with 2 mL of sulfuric acid (~1760 g/L) TS until white fumes are evolved, add drop by drop nitric acid (~1000 g/L) TS until oxidation is complete, and cool. Add 2 mL of water, heat until white fumes are again evolved, cool, add 10 mL of water and neutralize with ammonia (~100 g/L) TS using pH-indicator paper R. Keep half of the solution for test F. The remaining solution yields reaction A described under 2.1 General identification tests as characteristic of orthophosphates.
-
The solution prepared in test E yields reaction B described under 2.1 General identification tests as characteristic of sodium.
Specific optical rotation. Use a 10.0 mg/mL solution of the test substance in water R and calculate with reference to the anhydrous and ethanol-free substance; 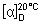 = +75 to +83.
= +75 to +83.
Clarity of solution. A solution of 0.50 g of the test substance in 10 mL of carbon-dioxide-free water R is clear.
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, method A, using about 0.200 g of the substance. The water content is not more than 100 mg/g.
Inorganic phosphates. Dissolve 0.050 g in sufficient water to produce 100 mL. To 10 mL, add 5 mL of ammonium molybdate/vanadate TS, mix and allow to stand for 5 minutes; any yellow colour produced is not more intense than that of a reference solution prepared similarly using 10 mL of phosphate standard (5 µg/mL) TS.
Ethanol. Carry out the test as described under 1.14.1 Chromatography, Gas chromatography with the apparatus equipped with an injection system for the performance of static head-space chromatography. Use a fused-silica capillary or wide-bore column 30 m long and 0.32 mm or 0.53 mm in internal diameter coated with macrogol 20 000 R (film thickness: 0.25 µm).
As a detector, use a flame ionization detector.
Use nitrogen for chromatography R or helium for chromatography R as the carrier gas at an appropriate pressure and a split ratio 1:5 with a linear velocity of about 35 cm/sec.
The following head-space injection conditions may be used:
|
Equilibration temperature (°C) |
80 |
|
Equilibration time (min) |
60 |
|
Transfer line temperature (°C) |
85 |
|
Pressurization time (s) |
30 |
|
Injection volume (mL) |
1 |
Maintain the temperature of the column at 30 °C for 7 minutes then raise the temperature at a rate of 35 °C per minute to 180 °C and maintain for 10 minutes, maintaining the temperature of the injection port at 140 °C and that of the flame ionization detector at 250 °C.
Test solution. Dissolve 0.200 g of the test substance in water R and dilute to 20.0 mL with the same solvent. Introduce 5.0 mL of this solution and 1.0 mL of water R into a headspace vial. Prepare two more vials.
Reference solutions. Add 0.100 g of ethanol R to water R and dilute to 200.0 mL with the same solvent. Transfer respectively 2.0 mL, 4.0 mL and 6.0 mL in separate headspace injection vials and bring the volume to 6.0 mL with water R, if necessary.
Blank solution. Introduce 6.0 mL of water R into a headspace vial.
Analyse the blank solution and then alternate 3 times the test solution and the 3 reference solutions.
The test is not valid unless the relative standard deviation on the areas of the peaks obtained from the test solutions is not more than 5%.
Calculate the ethanol content by using the results obtained with the test solution and with the reference solutions; the ethanol content is not more than 30 mg/g.
pH value ( 1.13 ). pH of a 10 mg/mL solution in carbon-dioxide-free water R, 7.5–9.5.
Related substances. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (12.5 cm × 4.6 mm) packed with end-capped, base-deactivated particles of silica gel, the surface of which has been modified with chemically-bonded octylsilyl groups (5 µm).
Prepare solution (A) by dissolving 7.0 g of ammonium acetate R in 1000 mL of water R.
The mobile phase for the gradient elution consists of a mixture of mobile phase A and mobile phase B using the following conditions:
- Mobile phase A : mix 30 volumes of solution (A) with 35 volumes of water R, adjust to pH 3.8 with glacial acetic acid R, then add 35 volumes of methanol R;
- Mobile phase B : mix 30 volumes of solution (A), adjusted to pH 4.0 with glacial acetic acid R and 70 volumes of methanol R.
|
Time (minutes) |
Mobile phase A |
Mobile phase B |
Comments |
|
0–3.5 |
90 |
10 |
Isocratic |
|
3.5–23.5 |
90 to 60 |
10 to 40 |
Linear gradient |
|
23.5–34.5 |
60 to 5 |
40 to 95 |
Linear gradient |
|
34.5–50 |
5 |
95 |
Isocratic |
|
50–55 |
5 to 90 |
95 to 10 |
Return to initial composition |
|
55–65 |
90 |
10 |
Re-equilibration |
Operate with a flow of 1.0 mL/min. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 254 nm. Maintain the column temperature at 30 °C.
Prepare the following solutions in mobile phase A. For solution (1), use 1.0 mg of the test substance per mL. For solution (2), use a solution containing 20 µg of betamethasone sodium phosphate RS per mL and 20 µg of dexamethasone sodium phosphate RS per mL. For solution (3), mix equal volumes of solution (2) and a solution containing 20 µg of dexamethasone RS per mL. For solution (4), dilute a suitable volume of solution (1) to obtain a concentration of 1 µg of dexamethasone sodium phosphate per mL.
Inject 20 µL of solution (2). The test is not valid unless the resolution between the peaks due to dexamethasone phosphate (retention time about 22 min) and betamethasone phosphate (with a relative retention of about 0.95) is at least 2.0.
Inject alternately 20 µL each of solutions (1), (3) and (4). In the chromatogram obtained with solution (3), the following peaks are eluted at the following relative retention with reference to dexamethasone phosphate (retention time about 22 min): impurity B (betamethasone phosphate): about 0.95; impurity A (dexamethasone): about 1.37. The chromatogram obtained with solution (1) may show the following impurities, in addition to impurity A and impurity B, at the following relative retention with reference to dexamethasone phosphate: impurity C: about 0.5; impurity D: about 0.6; impurity E: about 0.8; impurity F: about 0.92; impurity H: about 1.19; and impurity G: about 1.41.
In the chromatogram obtained with solution (1):
-
the area of any peak corresponding to impurity A, when multiplied by a correction factor of 0.75, is not greater than 5 times the area of the principal peak obtained with solution (4) (0.5%);
-
the area of any peak corresponding to impurity G is not greater than 3 times the area of the principal peak obtained with solution (4) (0.3 %);
-
the area of any peak corresponding to either impurity B, C, D, E or F is not greater than 2 times the area of the principal peak obtained with solution (4) (0.2%);
-
the area of any other peak, other than the principal peak, is not greater than the area of the principal peak obtained with solution (4) (0.10%);
-
the sum of the corrected area of any peak corresponding to impurity A and the areas of all other peaks, other than the principal peak, is not greater than 10 times the area of the principal peak obtained with the solution (4) (1.0 %). Disregard any peak with an area less than 0.05 times the area of the principal peak obtained with solution (4) (0.05%).
Assay
- Either method A or B may be applied.
-
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (15 cm × 4.6 mm) packed with end-capped, base-deactivated particles of silica gel, the surface of which has been modified with chemically-bonded octylsilyl groups (7 µm).
Prepare the mobile phase by mixing 520 mL of water R with 2 mL of phosphoric acid (~1440 g/L) TS. Adjust the temperature to 20 °C, then adjust to pH 2.6 with sodium hydroxide R. Mix this solution with 36 mL of tetrahydrofuran R and 364 mL of methanol R.
Operate with a flow of 1.5 mL/min. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 254 nm.
Prepare the following solutions in mobile phase unless otherwise stated. For solution (1), dissolve 30.0 mg of the test substance and dilute to 50.0 mL. Dilute 5.0 mL of this solution to 50.0 mL. For solution (2), dissolve 30.0 mg of dexamethasone phosphate RS and dilute to 50.0 mL. Dilute 5.0 mL of this solution to 50.0 mL. For solution (3), dissolve 2 mg of dexamethasone RS and 2 mg dexamethasone phosphate RS in 2 mL of tetrahydrofuran R, then dilute to 100.0 mL. Dilute 5.0 mL of this solution to 50.0 mL.
Inject solution (3). Record the chromatograms for about 3 times the retention time of dexamethasone phosphate. The assay is not valid unless the resolution factor between the two principal peaks due to dexamethasone phosphate (retention time about 8 minutes) and the peak due to dexamethasone (impurity A) (with a relative retention of about 2) is at least 6.0.
Inject alternately 20 µL each of solutions (1) and (2).
Measure the areas of the peaks corresponding to dexamethasone phosphate obtained and calculate the percentage content of dexamethasone sodium phosphate (C22H28FNa2O8P) using the declared content of dexamethasone phosphate C22H30FO8P in dexamethasone phosphate RS. Each mg of dexamethasone phosphate C22H30FO8P corresponds to 1.093 mg of dexamethasone sodium phosphate C22H28FNa2O8P.
-
Dissolve about 0.2 g, accurately weighed, in sufficient water to produce 200 mL. Dilute 5 mL to 250 mL with water and measure the absorbance of this solution (1.6) in a 1 cm layer at the maximum at about 241 nm. Calculate the content of Dexamethasone sodium phosphate (C22H28FNa2O8P) using the absorptivity value of 29.7 (
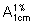 = 297).
= 297).
Additional requirements for Dexamethasone sodium phosphate for parenteral use.
Complies with the monograph for Parenteral preparations.
Bacterial endotoxins. Carry out the test as described under 3.4 Test for bacterial endotoxins; contains not more than 31.3 IU of endotoxin per mg.
Impurities
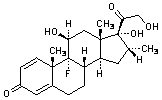
A. 9-fluoro-11β,17,21-trihydroxy-16α-methylpregna-1,4-diene-3,20-dione (dexamethasone).
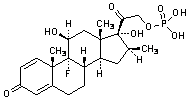
B. 9-fluoro-11β,17-dihydroxy-16β-methyl-3,20-dioxopregna-1,4-dien-21-yl dihydrogen phosphate (betamethasone phosphate).
 or
or 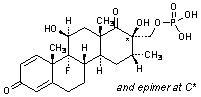
C, D, E, F. For each impurity, one or more diastereoisomer(s) of:
-
(9-fluoro-11β,17a-dihydroxy-16-methyl-3,17-dioxo-17a-homoandrosta-1,4-dien-17a-yl)methyl dihydrogen phosphate (undefined stereochemistry at C-16 and C-17a); or
-
(9-fluoro-11β,17-dihydroxy-16α-methyl-3,17a-dioxo-17a-homoandrosta-1,4-dien-17-yl)methyl dihydrogen phosphate (undefined stereochemistry at C-17).

G. 9-fluoro-11β,17-dihydroxy-16α-methyl-3-oxoandrosta-1,4-diene-17β-carboxylic acid.

H. 9-fluoro-11β,17-dihydroxy-16β-methyl-3,20-dioxopregn-4-en-21-yl dihydrogen phosphate.