Monographs: Pharmaceutical substances: Chlorhexidine digluconate solution (Chlorhexidini digluconatis solutio)
Molecular formula. C22H30Cl2N10,2C6H12O7
Relative molecular mass . 897.8
Graphic formula
Chemical name. 1,1'-(hexamethylene)bis[5-(4-chlorophenyl)biguanide] di-d-gluconate, 1,1'-(hexane-1,6-diyl)bis[5-(4-chlorophenyl)biguanide] di-d-gluconate, d-gluconic acid compd. with N1,N14-bis(4-chlorophenyl)-3,12-diimino-2,4,11,13-tetraazatetradecanediimidamide (2:1); CAS Reg. No. 18472-51-0.
Description. A clear, colourless or pale yellow liquid.
Miscibility. Miscible with water, with not more than 3 parts of acetone R and with not more than 5 parts of ethanol (~750 g/L) TS.
Category. Antiseptic.
Storage. Chlorhexidine digluconate solution should be kept in a well-closed container (avoid unlined steel containers), protected from light.
Requirements
Definition. Chlorhexidine digluconate solution is an aqueous solution of chlorhexidine digluconate. It contains not less than 190 g and not more than 210 g of chlorhexidine digluconate (C22H30Cl2N10,2C6H12O7) per L.
Identity tests
- Either tests A and B or tests B, C and D may be applied.
A. To 1 mL of the test solution add 40 mL of water R, cool in iced water, make alkaline to titan yellow paper R by adding dropwise, and with stirring, sodium hydroxide (~420 g/L) TS and add 1 mL in excess. Filter, wash the precipitate with water R until the washings are free from alkali. Recrystallize from methanol R. Dry the crystals at 100–105 ºC for 1 hour. Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from chlorhexidine RS or with the reference spectrum of chlorhexidine. If the spectra thus obtained are not concordant, repeat the test using the residues obtained by separately dissolving the test substance and chlorhexidine RS in a small amount of methanol R and evaporating to dryness. The infrared absorption spectrum is concordant with the spectrum obtained from chlorhexidine RS.
B. Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography using silica R5 as the coating substance and a mixture of 10 volumes of ammonia (~260 g/L) TS, 10 volumes of ethyl acetate R, 30 volumes of water R and 50 volumes of ethanol (~750 g/L) TS as the mobile phase. Apply separately to the plate 5 µL of each of the following 2 solutions in water R. For solution (A) dilute 10 mL of the test solution to a final volume of 50 mL. For solution (B) use 20 mg of calcium gluconate R per mL. After removing the plate from the chromatographic chamber dry the plate at 100 ºC for 20 minutes and allow to cool.
Spray the plate with a solution containing 25 g/L ammonium molybdate R and 10 g/L ceric sulfate R in sulfuric acid (~98 g/L) TS. Heat the plate for about 10 minutes at 110 °C. Examine the chromatogram in daylight.
The principal spot obtained with solution (A) corresponds in position, appearance and intensity to that obtained with solution (B).
C. Dissolve 1 mL of the test solution in 10 mL of water R and add, with shaking, 0.15 mL of copper (II) chloride/ammonia TS; a purple precipitate is produced immediately. Continue to add 0.5 mL of copper(II)chloride/ammonia TS; the colour of the precipitate changes to blue.
D. Carry out the test as described under 1.14.4 High-performance liquid chromatography using the conditions given under “Related substances”. The retention time of the principal peak in the chromatogram obtained with solution (1) is similar to that of the principal peak in the chromatogram obtained with solution (3).
pH value (1.13). Dilute 1 mL of the test solution to 20 mL in carbon-dioxide-free water R, 5.5–7.0.
Relative density (1.3).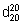 = 1.06 –1.07.
= 1.06 –1.07.
Chloroaniline
Prepare sample solution (A) by diluting 0.20 g of the test solution to 30 mL with water R. Add rapidly and with thorough mixing after each addition: 5 mL of hydrochloric acid (~103 g/L) TS, 0.35 mL of sodium nitrite (100 g/L) TS, 2 mL of ammonium sulfamate (50 g/L) TS, 5 mL of N-(1-naphthyl) ethylenediamine hydrochloride (1 g/L) TS and 1 mL of ethanol (~750 g/L) TS. Transfer this solution quantitatively to a 50.0 mL volumetric flask, dilute to volume with water R and allow to stand for 30 minutes.
Prepare reference solutions (B)–(F) representing respectively 0.005% (m/m), 0.01% (m/m), 0.02% (m/m), 0.05% (m/m) and 0.06% (m/m) of chloroaniline in the test solution as follows: Dilute 1.0 mL, 2.0 mL, 4.0 mL, 10.0 mL and 12.0 mL of a solution containing 10 µg per mL of chloroaniline R in hydrochloric acid (200 g/L) TS to 20 mL with water R. Then add 10.0 mL of water R. Add rapidly and with thorough mixing after each addition: 5 mL of hydrochloric acid (~103 g/L) TS, 0.35 mL of sodium nitrite (100 g/L) TS, 2 mL of ammonium sulfamate (50 g/L) TS, 5 mL of N-(1-naphthyl) ethylenediamine hydrochloride (1 g/L) TS and 1 mL of ethanol (~750 g/L) TS. Transfer each solution quantitatively to a 50.0 mL volumetric flask, dilute to volume with water R and allow to stand for 30 minutes.
Measure the absorbance (1.6) of solutions (A)–(F) in a 1 cm layer at the maximum at about 556 nm. Plot a calibration curve for solutions (B)–(F). Determine the concentration of chloroaniline from the calibration curve.
The content of chloroaniline is not more than 0.05% (m/m) of chloroaniline in the the chlorhexidine gluconate solution under investigation.
Related substances
Prepare fresh solutions and perform the tests without delay.
Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (25 cm × 4.6 mm) packed with particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups (5 µm). Maintain the column at 30 °C.
Use the following conditions for gradient solution:
mobile phase A: mix 20 volumes of a 0.1% (v/v) solution of trifluoroacetic acid R in acetonitrile R and 80 volumes of a 0.1% (v/v) solution of trifluroacetic acid R in water R;
mobile phase B: mix 10 volumes of a 0.1% (v/v) solution of trifluoroacetic acid R in water R and 90 volumes of a 0.1% (v/v) solution of trifluoroacetic acid R in acetonitrile R.
Use the following gradient elution:
|
Time |
Mobile phase A |
Mobile phase B |
Comments |
|
0–2 |
100 |
0 |
Isocratic |
|
2–32 |
100 to 80 |
0 to 20 |
Linear gradient |
|
32–37 |
80 |
20 |
Isocratic |
|
37–47 |
80 to 70 |
20 to 30 |
Linear gradient |
|
47–54 |
70 |
30 |
Isocratic |
|
54–56 |
70 to 100 |
30 to 0 |
Return to initial composition |
|
56–66 |
100 |
0 |
Re-equilibration |
Prepare the following solutions in mobile phase A: for solution (1) transfer 1.0 mL of the test solution to a 100 mL volumetric flask and dilute to volume. For solution (2) transfer 1.0 mL of solution (1) to a 100 mL volumetric flask and dilute to volume. For solution (3) dissolve the contents of a vial of chlorhexidine for system suitability RS (containing chlorhexidine and the impurities A, B, F, G, H, I, J, K, L, N and O) in 1.0 mL.
Operate with a flow rate of 1.0 mL per minute. As a detector use an ultraviolet spectrophotometer set at a wavelength of 254 nm.
Inject alternately 10 µL each of solutions (1), (2) and (3).
Use the chromatogram supplied with chlorhexidine for system suitability RS and the chromatogram obtained with solution (3) to identify the peaks due to impurities A, B, F, G, H, I, J, K, L, N and O.
Relative retention with reference to chlorhexidine (retention time = about 35 min):
impurity L = about 0.23; impurity Q = about 0.24; impurity G = about 0.25;
impurity N = about 0.35; impurity B = about 0.36; impurity F = about 0.5;
impurity A = about 0.6; impurity H = about 0.85; impurity O = about 0.90;
impurity I = about 0.91; impurity J = about 0.96; and impurity K = about 1.4.
The test is not valid unless in the chromatogram obtained with solution (3):
- the resolution between the peaks due to impurities L and G is at least 3.0;
- the peak-to-valley ratio (Hp/Hv) is at least 2.0, where Hp = height above the baseline of the peak due to impurity B and Hv = the height above the baseline of the lowest point of the curve separating this peak from that due to impurity N.
In the chromatogram obtained with solution (1):
- the area of any peak corresponding to impurity N is not greater than the area of the principal peak in the chromatogram obtained with solution (2) (1.0%);
- the area of any peak corresponding to impurity H is not greater than 0.5 times the area of the principal peak in the chromatogram obtained with solution (2) (0.5%);
- the areas of any peak corresponding to either impurities A, J or K is not greater than 0.4 times the area of the principal peak in the chromatogram obtained with solution (2) (0.4%);
- the sum of the areas of any peaks corresponding to impurities I and O is not greater than 0.4 times the area of the principal peak in the chromatogram obtained with solution (2) (0.4%);
- the area of any peak corresponding to impurity G is not greater than 0.3 times the area of the principal peak in the chromatogram obtained with solution (2) (0.3%);
- the areas of any peak corresponding to either impurities B, F, L or Q is not greater than 0.2 times the area of the principal peak in the chromatogram obtained with solution (2) (0.2%);
- the area of any other impurity peak is not greater than 0.1 times the area of the principal peak in the chromatogram obtained with solution (2) (0.10%);
- the sum of the areas of all impurity peaks is not greater than 3 times the area of the principal peak in the chromatogram obtained with solution (2) (3.0%). Disregard any peak with an area less than 0.05 times the area of the principal peak in the chromatogram obtained with solution (2) (0.05%).
Assay. Determine the weight per mL (1.3.1) of the test solution. Transfer about 1 g of the test solution, accurately weighed, to a 250 mL beaker and add 50 mL of anhydrous acetic acid R. Titrate with 0.1 M perchloric acid (0.1 mol/L) VS as described under 2.6 Non-aqueous titration, method A, determining the end-point potentiometrically.
Each mL of perchloric acid (0.1 mol/L) VS is equivalent to 22.44 mg of C34H54Cl2N10O14.
A. 1-(4-chlorophenyl)-5-{6-[(cyanocarbamimidoyl)amino]hexyl}biguanide
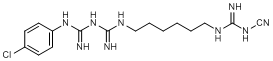
B. N-{[6-({[(4-chlorophenyl)carbamimidoyl]carbamimidoyl}amino)hexyl]carbamimidoyl}-urea
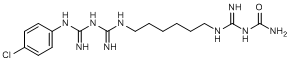
E. (4-chlorophenyl)guanidine
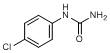
F. N-(4-chlorophenyl)urea
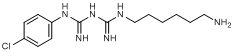
G. 1-(6-aminohexyl)-5-(4-chlorophenyl)biguanide
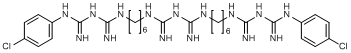
H. 1,1'-[iminobis(carbonimidoyliminohexamethylene)]bis[5-(4-chlorophenyl)biguanide]
I. unknown structure
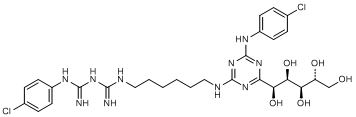
J. 1-(4-chlorophenyl)-5-[6-({4-[(4-chlorophenyl)amino]-6-[(1S,2R,3R,4R)-1,2,3,4,5-pentahydroxypentyl]-1,3,5-triazin-2-yl}amino)hexyl]biguanide
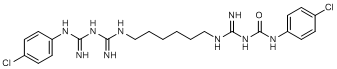
K. N-(4-chlorophenyl)-N'-{[6-({4-[(4-chlorophenyl)carbamimidoyl]carbamimidoyl}amino)hexyl]carbamimidoyl}urea
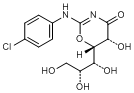
L. (5R,5S)-2-[(4-chlorophenyl)amino]-5-hydroxy-6-[(1R,2R)-1,2,3-trihydroxypropyl]-5,6-dihydro-4H-1,3-oxazin-4-one
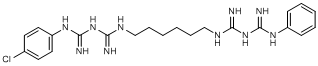
M. 1-(4-chlorophenyl)-5-(6-{[(phenylcarbamimidoyl)carbamimidoyl]amino}hexyl)biguanide
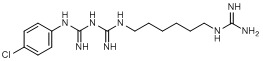
N. 11-[6-(carbamimidoylamino)hexyl]-5-(4-chlorophenyl)biguanide
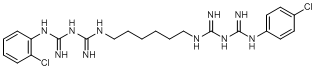
O. 1-(2-chlorophenyl)-5-[6-({[(4-chlorophenyl)carbamimidoyl]carbamimidoyl}amino)hexyl]biguanide
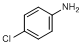
P. 4-chloroaniline
Q. unknown structure