Monographs: Pharmaceutical substances: Oseltamivir phosphate (Oseltamiviri phosphas)
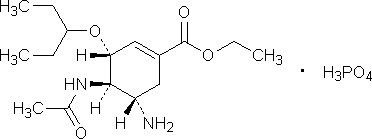
C16H28N2O4, H3PO4
Relative molecular mass. 410.4
Chemical name. Ethyl (3R,4R,5S)-4-(acetylamino)-5-amino-3-(1-ethylpropoxy)cyclohex-1-ene-1-carboxylate dihydrogen phosphate. CAS Reg. No. 204255-11-8.
Description. A white to off-white powder.
Solubility. Freely soluble in water and in methanol.
Category. Antiviral.
Storage. Oseltamivir phosphate should be kept in a well-closed container, protected from light.
Additional information. Oseltamivir phosphate may show polymorphism.
Requirements
Definition. Oseltamivir phosphate contains not less than 97.5% and not more than 102.0% of oseltamivir phosphate (C16H28N2O4, H3PO4) using Assay method A, and not less than 98.5% and not more than 101.0% of oseltamivir phosphate (C16H28N2O4, H3PO4) using Assay method B, both calculated with reference to the anhydrous substance.
Manufacture. The production method is validated to ensure that the substance is the (3R,4R,5S)-enantiomer and that less than 100 ppm of the impurity ethyl-(1R, 2R,3S,4R,5S)-4-acetamido-5-amino-2-azido-3-(1-ethylpropoxy)cyclohexanecarboxylate is present, when determined by a suitable method such as liquid chromatography combined with mass spectrometry (LC-MS). Where necessary, the production method is also validated to demonstrate that not more than 0.1% of tributyl-phosphine oxide is present in the final product, when examined by a suitable method such as gas chromatography (GC).
Identity test
• Either tests A, B and D or tests C and D may be applied.
A. Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography, using silica gel R6 as the coating substance and a mixture of 8 volumes of methanol R, 6 volumes of ethyl acetate R, 4 volumes of toluene R and 2 volumes of ammonia (~260g/l) TS as the mobile phase. Apply separately to the plate 10 µl of each of the following two solutions in methanol R, containing (A) 2 mg of the test substance per mL and (B) 2 mg of oseltamivir phosphate RS per mL. After removing the plate from the chromatographic chamber, allow it to dry exhaustively in air or in a current of cool air. Examine the chromatogram in ultraviolet light (254 nm).
The principal spot obtained with solution A corresponds in position, appearance, and intensity to that obtained with solution B.
B. Determine the specific optical rotation (1.4) using a 10 mg/mL solution and calculate with reference to the anhydrous substance;
= -30.7° to -32.6°.
C. Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from oseltamivir phosphate RS or with the reference spectrum of oseltamivir phosphate.
D. Neutralize a 4 mg/mL solution with a few mL of sodium hydroxide (0.1 mol/l) VS. Use 5 mL of this solution; it yields reaction B described under 2.1 General identification tests as characteristic of orthophosphates.
Heavy metals. Use 1.0 g for the preparation of the test solution as described under 2.2.3 Limit test for heavy metals, Procedure 1 and determine the heavy metal content according to Method A; not more than 10 µg/g.
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A. Use 1.0 g of the test substance. The water content is not more than 5 mg/g.
Related substances. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using the same conditions as under Assay, method A using solutions (1), (3) and (4).
Inject separately 15 μl each of solution (1), (3) and (4) and of the dissolution solvent in the chromatographic system. Examine the blank chromatogram for any extraneous peaks and disregard the corresponding peaks observed in the chromatogram obtained with solution (1).
Use the chromatogram obtained with solution (4) to identify the peaks due to impurities A, B, C and D. The impurity peaks are eluted at the following relative retention with reference to oseltamivir phosphate (retention time about 19 minutes): impurity A about 0.16, impurity B about 0.17, impurity C about 0.51 and impurity D about 0.55. The test is not valid unless the resolution between the peaks due to impurities A and B and that between the peaks due to impurities C and D is at least 1.3.
In the chromatogram obtained with solution (1) the area of any peak corresponding to impurity B, when multiplied by a correction factor of 1.4, is not greater than 3 times the area of the peak in the chromatogram obtained with solution (3) (0.3%), the area of any peak corresponding to impurity C, when multiplied by a correction factor of 0.6, is not greater than 1.5 times the area of the peak in the chromatogram obtained with solution (3) (0.15%), the area of any other peak, apart from the principal peak, is not greater than the area of the peak in the chromatogram obtained with solution (3) (0.1%). The sum of the corrected areas of any peaks corresponding to impurities B or C and of the areas of all other peaks, apart from the principal peak, is not greater than 7 times the area of the peak obtained with solution (3) (0.7%). Disregard any peak with an area less than 0.5 times the area of the principal peak obtained with solution (3) (0.05%).
Assay
• Either method A or B may be applied.
A. Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography, using a stainless steel column (25 cm x 4.6 mm) packed with octylsilyl silica gel for chromatography (5 µm).
The mobile phase consists of a mixture of 620 volumes of 0.05 M potassium dihydrogen phosphate (adjusted to pH 6 with potassium hydroxide (~110g/l TS), 245 volumes methanol R and 135 volumes acetonitrile R.
Operate with a flow rate of 1.2 mL per minute and the column oven temperature at 50°C. As a detector use an ultraviolet spectrophotometer set at a wavelength of about 207 nm.
Prepare the following solutions in a mixture of 620 volumes of water R, 245 volumes of methanol R and 135 volumes of acetonitrile R( dissolution solvent).
For solution (1) dissolve 50 mg of the test substance in the dissolution solvent and dilute to 50.0 mL with the same solvent. For solution (2) dissolve 50 mg of oseltamivir phosphate RS in the dissolution solvent and dilute to 50.0 mL with the same solvent. For solution (3) dilute 1.0 mL of solution (1) to 100 mL with dissolution solvent and then dilute 1.0 mL of this solution to 10 mL with the same solvent. For solution (4) dissolve about 2 mg of oseltamivir for system suitability RS in the dissolution solvent and dilute to 2 mL with the same solvent.
Inject separately 15 µl each of solution (1), (2) and (4) and of the dissolution solvent in the chromatographic system. The assay is not valid unless, in the chromatogram obtained with solution (4), the resolution between the peaks due to impurities A and B and that between the peaks due to impurities C, and D is at least 1.3.
Measure the areas of the peak responses in the chromatograms obtained with solutions (1) and (2). Calculate the percentage content of oseltamivir (C16H28N2O4, H3PO4) with reference to the anhydrous substance.
B. Dissolve about 0.300 g of oseltamivir phosphate, accurately weighed, in 30 mL of anhydrous acetic acid R and titrate with perchloric acid (0.1 mol/l) VS, determining the end-point potentiometrically as described under 2.6 Non-aqueous titration, Method A.
Each mL of perchloric acid (0.1 mol/l) VS is equivalent to 41.04 mg of (C16H28N2O4, H3PO4).
Impurities
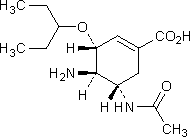
A. (3R,4R,5S)-5-(acetylamino)-4-amino-3-(1-ethylpropoxy)cyclohex-1-ene-1-carboxylic acid
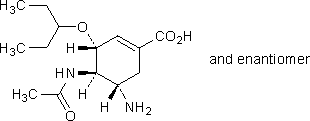
B. (3RS,4RS,5SR)-4-(acetylamino)-5-amino-3-(1-ethylpropoxy)cyclohex-1-ene-1-carboxylic acid,
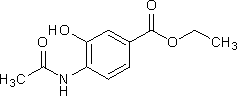
C. ethyl 4-(acetylamino)-3-hydroxybenzoate,
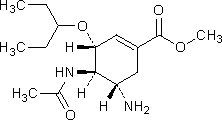
D. methyl (3R,4R,5S)-4-(acetylamino)-5-amino-3-(1-ethylpropoxy)cyclohex-1-ene-1-carboxylate,
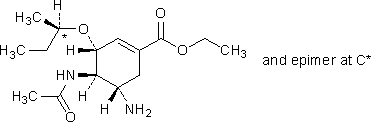
E. ethyl (3R,4R,5S)-4-(acetylamino)-5-amino-3-[(1RS)-1-methylpropoxy)cyclohex-1-ene-1-carboxylate,
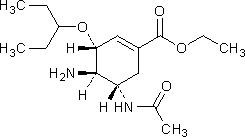
F. ethyl (3R,4R,5S)-5-(acetylamino)-4-amino-3-(1-ethylpropoxy)cyclohex-1-ene-1-carboxylate.