Monographs: Pharmaceutical substances: Daclatasvir dihydrochloride (Daclatasviri dihydrochloridum)
Molecular formula. C40H50N8O6.2HCl
Relative molecular mass. 811.8
Graphic formula.
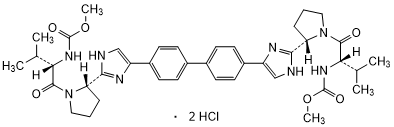
Chemical name. Dimethyl N,N'-([1,1'-biphenyl]-4,4'-diylbis{1H-imidazole-4,2-diyl-[(2S)-pyrrolidine-2,1-diyl] [(2S)-3-methyl-1-oxobutane-1,2-diyl]})dicarbamate dihydrochloride (IUPAC), Carbamic acid, N,N'-[[1,1'-biphenyl]-4,4'-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C'-dimethyl ester (CAS); CAS Reg. No. 1009119-65-6
Description. A white to pale yellow powder.
Solubility. Freely soluble in water, soluble in methanol and very slightly soluble in dimethylformamide.
Category. Antiviral (hepatitis C virus nonstructural protein 5A inhibitor).
Storage. Daclatasvir dihydrochloride should be kept in a tightly closed container.
Additional information. Daclatasvir dihydrochloride may exhibit polymorphism. The substance is slightly hygroscopic.
Requirements
Manufacture. The production method is validated to demonstrate that genotoxic halogenated biphenyl derivatives are adequately controlled in the final product.
Definition. Daclatasvir dihydrochloride contains not less than 97.0% and not more than 102.0% (“Assay”, Method A) or not less than 99.0% and not more than 101.0% (“Assay”, Method B) of C40H50N8O6.2HCl, calculated with reference to the anhydrous substance.
Identity tests.
- Either tests A, D and E or tests D and E, together with any one of tests B or C, may be applied.
A. Carry out the examination as described under 1.7 Spectrophotometry in the infrared region. The infrared absorption spectrum is concordant with the spectrum obtained from daclatasvir dihydrochloride RS or with the reference spectrum of daclatasvir dihydrochloride.
If the spectra thus obtained are not concordant, repeat the test using the residues obtained by separately dissolving the test substance and daclatasvir dihydrochloride RS in a small amount of methanol R and evaporating to dryness. The infrared absorption spectrum is concordant with the spectrum obtained from daclatasvir dihydrochloride RS.
B. Carry out text B.1 or, where a diode array detector is available, test B.2.
B.1 Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions given under “Assay”, Method A. The retention time of the principal peak in the chromatogram obtained with solution (1) corresponds to the retention time of the peak due to daclatasvir in the chromatogram obtained with solution (2).
The absorption spectrum (1.6) of a 10 µg per mL solution of the test substance in methanol R, when observed between 230 nm and 400 nm, exhibits one maximum at 314 nm.
B.2 Carry out the test as described under 1.14.4 High-performance-liquid chromatography using the conditions given under “Assay”, Method A. Record the UV spectrum of the principal peak in the chromatograms with a diode array detector in the range of 230 nm to 400 nm. The retention time and the UV spectrum of the principal peak in the chromatogram obtained with solution (1) correspond to the retention time and the UV spectrum of the peak due to daclatasvir in the chromatogram obtained with solution (2).
C. Carry out the test as described under 1.14.1 Chromatography, Thin-layer chromatography using silica gel R4 or similar as the coating substance and a mixture of 77 volumes of ethyl acetate R, 15 volumes of methanol R and 8 volumes of water R as the mobile phase. Apply separately to the plate 2 μL of each of the following two solutions in methanol R containing (A) 10 mg of the test substance per mL and (B) 10 mg of daclatasvir dihydrochloride RS per mL. After removing the plate from the chromatographic chamber, allow it to dry in air or in a current of cool air. Examine the chromatogram in ultraviolet light (365 nm). The principal spot obtained with solution (A) corresponds in position, appearance and intensity with that obtained with solution (B).
Dip the plate in modified Dragendorff reagent TS. Dry it and examine the chromatogram in daylight. The principal spot obtained with solution (A) corresponds in position, appearance and intensity with that obtained with solution (B).
D. Carry out test D.1 or, where HPLC and the indicated chiral column are available, test D.2.
D.1 Determine the specific optical rotation (1.4) using a 10 mg per mL solution of the test substance in methanol R and calculate with reference to the anhydrous substance:
= -92.0 to -102.0.
D.2 Carry out the test as described under 1.14.1 Chromatography, High-performance liquid chromatography using the conditions and solutions given under “Impurity A (daclatasvir enantiomer).” The retention time of the principal peak obtained with solution (1) corresponds to the retention time of the peak due to daclatasvir in the chromatogram obtained with solution (3).
E. Dissolve 20 mg of the test substance in 20 mL methanol R; the solution yields reaction A described under 2.1 General identification tests as characteristic of chlorides.
pH value. pH of a 10 mg/mL solution of the test substance in carbon-dioxide-free water R, 2.5-3.5.
Impurity A (daclatasvir enantiomer). Carry out test as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (15 cm x 4.6 mm) packed with particles of silica gel, the surface of which has been modified with chemically-bonded cellulose tris (3,5-dichlorophenyl carbamate) (3 µm). As mobile phase, use a mixture of 30 volumes of 1.58 g per litre ammonium bicarbonate R in water and 70 volumes of acetonitrile R.
Operate at a flow rate of 1.0 mL per minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 320 nm. Maintain the column temperature at 40 °C.
Prepare the following solutions in mobile phase. For solution (1), dissolve 25.0 mg of the test substance in 50.0 mL. For solution (2), dilute 5.0 mL of solution (1) to 100.0 mL. Dilute 2.0 mL of this solution to 100.0 mL. For solution (3), dissolve the content of a vial of daclatasvir for system suitability RS (containing daclatasvir and impurity A) in 1 mL.
Inject alternately 10 µL of solutions (1), (2) and (3).
The test is not valid unless, in the chromatogram obtained with solution (3), the peak-to-valley ratio (Hp/Hv) is at least 3.0, where Hp is the height above the extrapolated baseline of the peak due to impurity A and Hv is the height above the extrapolated baseline of the lowest point of the curve separating this peak from the peak due to daclatasvir. Impurity A is eluted with a relative retention of about 1.6 with reference to daclatasvir (retention time about 4.5 minutes). Also, the test is not valid unless in the chromatogram obtained with solution (2) the peak due to daclatasvir is detected with a signal-to-noise ratio of at least 10.
In the chromatogram obtained with solution (1):
- the area of any peak corresponding to impurity A is not greater than 1.5 times the area of the peak due to daclatasvir in the chromatogram obtained with solution (2) (0.15%).
Related substances. Carry out the test as described under 1.14.4 High-performance liquid chromatography using the conditions given under “Assay”, Method A with the following modifications:
Use the following conditions for gradient elution:
- mobile phase A: 0.1% (v/v) solution of trifluoroacetic acid R;
- mobile phase B: mixture of 30 volumes of methanol R and 70 volumes of acetonitrile R.
|
Time (minutes) |
Mobile phase A |
Mobile phase B |
Comments |
|
0–1 |
80 |
20 |
Isocratic |
|
1–25 |
80 to 55 |
20 to 45 |
Linear gradient |
|
25–30 |
55 to 30 |
45 to 70 |
Linear gradient |
|
30–35 |
30 |
70 |
Isocratic |
|
35–37 |
30 to 80 |
70 to 20 |
Return to initial composition |
|
37–45 |
80 |
20 |
Re-equilibration |
Prepare the following solutions using as diluent a mixture of 80 volumes of mobile phase A and 20 volumes of mobile phase B. For solution (1), dissolve 25.0 mg of the test substance and dilute to 50.0 mL. For solution (2), dilute 1.0 mL of solution (1) to 100.0 mL. For solution (3), dilute 5.0 mL of solution (2) to 50.0 mL. For solution (4), dissolve the content of a vial of daclatasvir for peak identification RS (containing daclatasvir and the impurities B, E, G, H, I and J) in 1 mL.
Inject alternately 10 µL of solutions (1), (2), (3) and (4).
Use the chromatograms supplied with daclatasvir for peak identification RS and obtained with solution (4) to identify the peaks due to the impurities B, E, G, H, I and J. The impurities, if present, are eluted at the following relative retentions with reference to daclatasvir (retention time about 17 minutes): impurity J about 0.21; impurity I about 0.62; impurity H about 0.76; impurities B and C about 1.12; impurity F about 1.16; impurities D and E about 1.22; impurity K about 1.39; impurity L about 1.66 and impurity G about 1.82.
The test is not valid unless in the chromatogram obtained with solution (4) the peak-to-valley ratio (Hp/Hv) is at least 20, where Hp is the height above the extrapolated baseline of the peak due to the co-eluting impurities B and C and Hv is the height above the extrapolated baseline at the lowest point of the curve separating the peak due to daclatasvir from the peak due to the co-eluting impurities B and C. Also, the test is not valid unless in the chromatogram obtained with solution (3) the peak due to daclatasvir is detected with a signal-to-noise ratio of at least 20.
In the chromatogram obtained with solution (1):
- the sum of the areas of any peaks corresponding to impurities B and C (impurities B and C co-elute) is not greater than 1.5 times the area of the peak due to daclatasvir obtained with solution (3) (0.15%);
- the sum of the areas of any peaks corresponding to impurities D and E (impurities D and E co-elute) is not greater than 1.5 times the area of the peak due to daclatasvir obtained with solution (3) (0.15%);
- the area of any peak corresponding to impurity J, when multiplied by a correction factor of 0.74, is not greater than 1.5 times the peak due to daclatasvir obtained with solution (3) (0.15%);
- the area of any peak corresponding to impurity G, when multiplied by a correction factor of 1.46, is not greater than 1.5 times the peak due to daclatasvir obtained with solution (3) (0.15%);
- the area of any peak corresponding to either impurities I or H is not greater than 1.5 times the area of the peak due to daclatasvir obtained with solution (3) (0.15%);
- the area of any other impurity peak is not greater than the area of the peak due to daclatasvir obtained with solution (3) (0.10%).
- The sum of the areas of all impurity peaks is not greater than the area of the principal peak obtained with solution (2) (1.0%). Disregard any peak with an area less than 0.5 times the area of the peak due to daclatasvir obtained with solution (3) (0.05%).
Water. Determine as described under 2.8 Determination of water by the Karl Fischer method, Method A, using 0.500 g of the substance; the water content is not more than 10 mg/g.
Heavy metals. Use 1.0 g for the preparation of the test solution as described under 2.2.3 Limit test for heavy metals, Procedure 1; determine the heavy metals content according to Method A; not more than 20 μg/g.
Sulfated ash (2.3). Not more than 1.0 mg/g.
Assay
- Either Method A or Method B may be applied.
A. Carry out test as described under 1.14.1 Chromatography, High-performance liquid chromatography using a stainless steel column (15 cm x 4.6 mm) packed with base-deactivated particles of silica gel, the surface of which has been modified with chemically-bonded octadecylsilyl groups (3.5 µm).
Use the following conditions for gradient elution:
o mobile phase A: 0.1 % (v/v) solution of trifluoroacetic acid R;
o mobile phase B: mixture of 50 volumes of methanol R and 50 volumes of acetonitrile R.
Time (minutes)
Mobile phase
A (% v/v)Mobile phase
B (% v/v)Comments
0–1
70
30
Isocratic
1–13
70 to 60
30 to 40
Linear gradient
13–16
60 to 15
40 to 85
Linear gradient
16–18
15
85
Isocratic
18–20
15 to 70
85 to 30
Return to initial composition
20–25
70
30
Re-equilibration
Operate at a flow rate of 1.0 mL/minute. As a detector, use an ultraviolet spectrophotometer set at a wavelength of 304 nm. For identity test B.2, use a diode array detector in the range of 230 nm to 400 nm. Maintain the column at a temperature of 40 °C.
Prepare the following solutions using, as diluent, a mixture of 70 volumes of mobile phase A and 30 volumes of mobile phase B.
For solution (1), dissolve 50.0 mg of the substance to be examined and dilute to 100.0 mL. Dilute 10.0 mL of this solution to 50.0 mL. For solution (2), dissolve 50.0 mg of daclatasvir dihydrochloride RS and dilute to 100.0 mL. Dilute 10.0 mL of this solution to 50.0 mL.
Inject alternately 20 µL each of solutions (1) and (2).
Measure the areas of the peak responses obtained in the chromatograms from solutions (1) and (2) and calculate the percentage content of daclatasvir dihydrochloride (C40H50N8O6·2HCl) using the declared content of C40H50N8O6·2HCl in daclatasvir dihydrochloride RS.
B. Dissolve 0.250 g in 10 mL water and add 40 mL of ethanol (~750 g/L) TS. Titrate with sodium hydroxide (0.1 mol/L) VS, determining the end-point potentiometrically. Each mL of sodium hydroxide (0.1 mol/L) VS is equivalent to 40.59 mg of C40H50N8O6·2HCl.
Impurities.
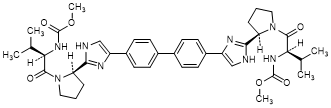
A. dimethyl N,N'-([1,1'-biphenyl]-4,4'-diylbis{1H-imidazole-4,2-diyl-[(2R)-pyrrolidine-2,1-diyl][(2R)-3-methyl-1-oxobutane-1,2-diyl]})dicarbamate (daclatasvir enantiomer) (synthesis-related impurity).
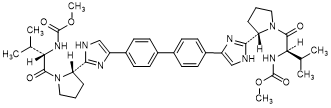
B. methyl N-[(2R)-1-{(2S)-2-[4-(4'-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methyl butanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}[1,1'-biphenyl]-4-yl)-1H-imidazol-2-yl] pyrrolidin-1-yl}-3-methyl-1-oxobutan-2-yl]carbamate (RS/SS-daclatasvir diastereoisomer) (synthesis-related impurity).
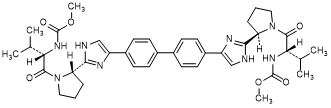
C. methyl N-[(2S)-1-{(2R)-2-[4-(4'-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methy lbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}[1,1'-biphenyl]-4-yl)-1H-imidazol-2-yl] pyrrolidin-1-yl}-3-methyl-1-oxobutan-2-yl]carbamate (SR/SS-daclatasvir diastereoisomer) (synthesis-related impurity).
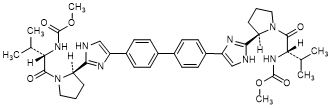
D. dimethyl N,N'-([1,1'-biphenyl]-4,4'-diylbis{1H-imidazole-4,2-diyl-[(2R)-pyrrolidine-2,1-diyl][(2S)-3-methyl-1-oxobutane-1,2-diyl]})dicarbamate (SR/SR-daclatasvir diastereoisomer) (synthesis-related impurity).
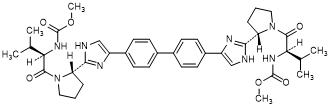
E. dimethyl N,N'-([1,1'-biphenyl]-4,4'-diylbis{1H-imidazole-4,2-diyl-[(2S)-pyrrolidine-2,1-diyl][(2R)-3-methyl-1-oxobutane-1,2-diyl]})dicarbamate (RS/RS-daclatasvir diastereoisomer) (synthesis-related impurity).
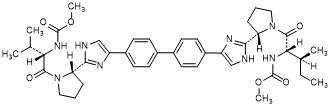
F. methyl N-[(2S,2S)-1-{(2S)-2-[4-(4'-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}[1,1'-biphenyl]-4-yl)-1H-imidazol-2-yl]pyrrolidin-1-yl}-3-methyl-1-oxopentan-2-yl]carbamate (synthesis-related impurity).
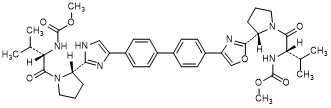
G. methyl N-[(2S)-1-{(2S)-2-[4-(4'-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}[1,1'-biphenyl]-4-yl)-1,3-oxazol-2-yl]pyrrolidin-1-yl}-3-methyl-1-oxobutan-2-yl]carbamate (synthesis-related impurity).
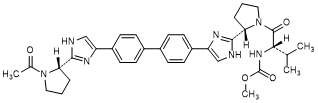
H. methyl N-[(2S)-1-{(2S)-2-[4-(4'-{2-[(2S)-1-acetylpyrrolidin-2-yl]-1H-imidazol-4-yl}[1,1'-biphenyl]-4-yl)-1H-imidazol-2-yl]pyrrolidin-1-yl}-3-methyl-1-oxobutan-2-yl]carbamate (synthesis-related impurity).
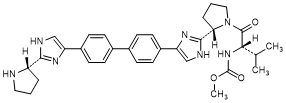
I. methyl N-[(2S)-1-{(2S)-2-[4-(4'-{2-[(2S)-pyrrolidin-2-yl]-1H-imidazol-4-yl}[1,1'-biphenyl]-4-yl)-1H-imidazol-2-yl]pyrrolidin-1-yl}-3-methylbutan-2-yl]carbamate (synthesis-related impurity).
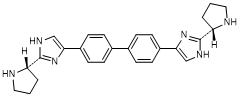
J. 4,4'-([1,1'-biphenyl]-4,4'-diyl)bis{2-[(2S)-pyrrolidin-2-yl]-1H-imidazole} (synthesis-related impurity).
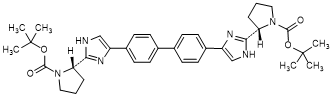
K. di-tert-butyl [1,1'-biphenyl]-4,4'-diylbis{1H-imidazole-4,2-diyl-[(2S)-pyrrolidine-1-carboxylate]} (synthesis-related impurity).
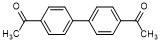
L. 1,1'-([1,1'-biphenyl]-4,4'-diyl)diethanone (synthesis-related impurity).